




Βλεννοπολυσακκχαριδώσεις (Mucopolysaccharidoses)
ΒΛΕΝΝΟΠΟΛΥΣΑΚΚΧΑΡΙΔΩΣΕΙΣ
1. ΒΛΕΝΝΟΠΟΛΥΣΑΚΚΧΑΡΙΔΩΣΗ ΤΥΠΟΥ Ι (Mucopolysaccharidosis type I; MPS I)
ΣΥΝΩΝΥΜΑ
- Ανεπάρκεια α-L-ιδουρονιδάσης (alpha-L-Iduronidase deficiency)
- Ανεπάρκεια IDUA (IDUA deficiency, MPS I)
ΕΠΙΔΗΜΙΟΛΟΓΙΑ
Συχνότητα. H MPS I απαντάται σε όλους τους πληθυσμούς σε συχνότητα περίπου 1:100,000 ζώντων παιδιών για τον βαρύ τύπο και 1:500,000, για τον ήπιο τύπο (Lowry RB et al, 1990; Meikle PJ et al, 1999; Poorthuis BJ et al, 1999). Ο ήπιος τύπος αποτελεί περίπου το 20% του συνολικού πληθυσμού των ασθενών με MPS I (Scott HS et al, 1995).
Στην Αγγλία και την Ουαλλία, η επίπτωση της MPS I στο χρονικό διάστημα 1981-2003 ήταν 1.07 περιπτώσεις/100.000 γεννήσεις (Moore D et al, 2008).
Φυλή. Η MPS I, επειδή κληρονομείται σύμφωνα με τον αυτοσωμικό υπολειπόμενο πρότυπο κληρονομικότητας, παρατηρείται εξίσου και στα 2 φύλα.
ΤΑΞΙΝΟΜΗΣΗ
Με βάση την κλινική παρουσίαση, η MPS I περιλαμβάνει 3 ξεχωριστά νοσήματα :
- Σύνδρομο Hurler (MPS IH) (βαρύ)
- Σύνδρομο Hurler-Scheie (MPS HIS) (ενδιάμεσο)
- Σύνδρομο Scheie (MPS IS) (ήπιο)
Επειδή όμως δεν έχουν καθορισθεί συγκεκριμένα κλινικά κριτήρια που να ξεχωρίζουν τα 3 αυτά σύνδρομα μεταξύ τους, ιδιαίτερα τα σύνδρομα Hurler-Scheie και Scheie, η MPS I, ανάλογα με την βαρύτητά της, έχει ταξινομηθεί σε 2 μεγαλύτερες ομάδες (Muenzer J et al, 2009) :
- Βαριά (severe) MPS I (σύνδρομο Hurler) και
- Hπια (attenuated) MPS I (σύνδρομα Hurler-Scheie και Scheie)
1.1 ΒΑΡΙΑ ΒΛΕΝΝΟΠΟΛΥΣΑΚΧΑΡΙΔΩΣΗ ΤΥΠΟΥ Ι (σύνδρομο Hurler) (severe MPS I; Hurler syndrome)
ΧΑΡΑΚΤΗΡΙΣΤΙΚΑ ΝΟΣΟΥ
Η βαριά MPS I χαρακτηρίζεται από χρόνια και προοδευτική διαδρομή, με προσβολή πολλαπλών οργανικών συστημάτων και ιστών (Clarke LA, 1997, Neufeld and Muenzer, 2001; Muenzer J et al, 2009). Η διάγνωσή της γίνεται κατά τον 9ο, κατά μέσον όρο, μήνα της ηλικίας. Τα περισσότερα πάσχοντα βρέφη ανακαλύπτονται πριν από τον 18ο μήνα της ηλικίας.
Τα βρέφη με βαριά MPS I φαίνονται φυσιολογικά στην γέννηση, αλλά, στη διάρκεια των 2 πρώτων ετών της ζωής, παρουσιάζουν τράχυνση των χαρακτηριστικών του προσώπου.
Τα πρώτα συμπτώματα (βουβωνοκήλες/ομφαλοκήλες, μακροκεφαλία, περιορισμός της απαγωγής των ισχίων, υποτροπιάζουσες αναπνευστικές λοιμώξεις) παρουσιάζονται στην διάρκεια του 1ου έτους της ζωής.
Ολοι οι ασθενείς με βαριά MPS I παρουσιάζουν προοδευτική δυσπλασία όλων των οστών (πολλαπλή δυσόστωση) και βαριά διανοητική αναπηρία. Κατά το 3ο έτος της ηλικίας, η γραμμική ανάπτυξη ανακόπτεται. Απώλεια της ακοής είναι συχνή, όπως συχνή είναι και η ανάπτυξη ύβου στο κατώτερο τμήμα της ΣΣ.
Οι τυπικές του εκδηλώσεις (περιλαμβανομένης της μακροκεφαλίας και της τράχυνσης των χαρακτηριστικών του προσώπου) παρουσιάζονται κατά το 12ο έτος της ηλικίας, ενώ ο περιορισμός της κινητικότητας και οι παραμορφώσεις των αρθρώσεων, μεταξύ 12ου-15ου έτους της ηλικίας (Schmidt H et al, 1987).
Αλλοτε παρουσιάζεται μόνο με αποτυχία της ανάπτυξης, θολερότητες του κερατοειδούς, ήπια τράχυνση των χαρακτηριστικών του προσώπου, ηπατομεγαλία και μικρογναθία (στην MPS I H/ S). Οι σκελετικές ανωμαλίες των χεριών και της ΣΣ είναι ελάχιστες. Σε μερικά βρέφη ηλικίας <1 έτους, η πρώτη εκδήλωση της νόσου είναι οξεία μυοκαρδιοπάθεια, συνδεόμενη με ενδοκαρδιακή ίνωση (Donaldson MDC et al, 1989).
Η κλινική εικόνα του συνδρόμου Hurler ολοκληρώνεται συνήθως κατά το 2ο έτος της ζωής. Ο θάνατος ενσκήπτει συνήθως στην διάρκεια της πρώτης δεκαετίας της ζωής, σαν αποτέλεσμα καρδιοαναπνευστικής ανεπάρκειας και προοδευτικής νευρολογικής προσβολής (Neufeld EF and Muenzer J, 2001).
ΑΙΤΙΟΛΟΓΙΑ
Το σύνδρομο Hurler κληρονομείται σύμφωνα με το αυτοσωμικό υπολειπόμενο πρότυπο κληρονομικότητας. Οφείλεται σε μεταλλάξεις του γονιδίου το οποίο κωδικοποιεί το λυσοσωμικό ένζυμο α –L-ιδουρονιδάση (IDUA; 252800) και έχει χαρτογραφηθεί στη χρωμοσωμική ταινία 4p16.3 (Banikazemi M, 2009). Στη χρωμοσωμική αυτή περιοχή έχουν ανευρεθεί πολλές διαφορετικές μεταλλάξεις, όπως αυτές που προκαλούν την MPS IH, MPS IS και MPS IH/S. Η ανεπάρκεια της α-L-ιδουρονιδάσης οδηγεί σε άθροιση μη αποδομημένων βλεννοπολυσακκχαριδών (θειική δερματάνη και κυρίως θειική ηπαράνη) σε διάφορα όργανα και ιστούς, οι οποίες ευθύνονται για την προοδευτική ανάπτυξη ορισμένων μορφολογικών ανωμαλιών. Παράλληλα, η θειική δερματάνη και η θειική ηπαράνη αποβάλλονται σε αυξημένες ποσότητες από τα ούρα.
ΚΛΙΝΙΚΕΣ ΕΚΔΗΛΩΣΕΙΣ
Γαστρεντερικό
- Ηπατοσπληνομεγαλία.
- Προβολή της κοιλιάς οφειλόμενη σε προοδευτική ηπατοπληνομεγαλία. Είναι συχνή σε ασθενείς με βαριά MPS I (Clarke LA, 1997). Πάντως, η εναπόθεση γλυκοζαμινογλυκανών στο ήπαρ και τον σπλήνα δεν προκαλεί δυσλειτουργία των οργάνων αυτών.
- Διάρροια. Για ανεξήγητους λόγους, πολλά παιδιά με βαριά MPS I παρουσιάζουν περιοδική απώλεια κοπράνων και διάρροια, ενίοτε εναλλασσόμενη με περιόδους σοβαρής δυσκοιλιότητας. Οι εκδηλώσεις αυτές μπορεί να περιορισθούν με την πάροδο του χρόνου ή να παροξυνθούν από μυική αδυναμία και έλλειψη φυσικών δραστηριοτήτων, όπως και από την χρήση αντιβιοτικών για άλλους λόγους (Clarke L and MacFarland J, 2001).
- Οι γαστρεντερικές διαταραχές στους ασθενείς με MPSs μπορεί να οφείλονται σε άτυπες γαστρεντερικές μικροβιακές λοιμώξεις (Wegrzyn G et al, 2005).
- Βουβωνοκήλες/ομφαλοκήλες.
Καρδιαγγειακό.
Η καρδιά προσβάλλεται σε όλους τους ασθενείς με MPS I. Η προσβολή της μπορεί είναι ανακαλυφθεί υπερηχοκαρδιογραφικά, πολύ ενωρίτερα πριν εκδηλωθεί κλινικά.
- Παλινδρόμηση μιτροειδούς και αορτής. Η προοδευτική πάχυνση και δυσκαμψία των φύλλων των καρδιακών βαλβίδων οδηγεί σε παλινδρόμηση της μιτροειδούς και της αορτής, η οποία μπορεί να γίνει αιμοδυναμικά σημαντική στα όψιμα στάδια της νόσου. Η παλινδρόμηση της μιτροειδούς είναι η συχνότερη βαλβιδοπάθεια στο σύνδρομο Hurler (Neufeld EF and Muenzer J, 2001).
- Ενδοκαρδιακή ινοελάστωση. Μερικοί ασθενείς με σύνδρομο Hurler στα πρώιμα στάδια της νόσου αναπτύσσουν ενδοκαρδιακή ινοελάστωση, η οποία μπορεί να οδηγήσει σε θανατηφόρα καρδιακή ανεπάρκεια (Stephan MJ et al, 1989)
- Αιφνίδιος θάνατος από αρρυθμία
- Καρδιαγγειακή κατέρειψη
- Μυοκαρδιοπάθεια
- Στεφανιαία αρτηριοπάθεια
Κρανιοπροσωπικές ανωμαλίες.
Η πάχυνση του θόλου του κρανίου οδηγεί σε μακροκεφαλία. Σκαφοκεφαλία είναι συχνή, λόγω πρόωρης σύγκλεισης των μετωπιαίων και εγκάρσιων ραφών.
Στην διάρκεια των 2 πρώτων ετών της νόσου, οι ασθενείς με σύνδρομο Hurler παρουσιάζουν τράχυνση των χαρακτηριστικών του προσώπου οφειλόμενη σε εναποθέσεις γλυκοζαμινογλυκανών στα μαλακά μόρια της στοματοπροσωπικής περιοχής σε έδαφος δυσόστωσης των οστών του προσώπου. Προοδευτικά ακόμα εμφανίζουν πάχυνση των ρινικών πτερυγίων, των χειλέων, των λοβών των ώτων και της γλώσσας.
Κατά το 2ο έτος της ηλικίας εμφανίζουν υπερτρίχωση του προσώπου και του σώματος. Οι τρίχες της κεφαλής γίνονται τραχιές, ευθειασμένες και παρόμοιες με «αχυροσκεπή».
Σκελετικές ανωμαλίες.
Κατά το 3ο έτος της ηλικίας η γραμμική ανάπτυξη ανακόπτεται. Δυσκαμψία των αρθρώσεων παρουσιάζεται συχνά κατά το 2ο έτος της ηλικίας.
Ολοι οι ασθενείς με σύνδρομο Hurler αναπτύσσουν προοδευτική αρθροπάθεια, η οποία οδηγεί σε σοβαρές αρθρικές παραμορφώσεις και γαμψοδακτυλία, λόγω φαλαγγικής δυσόστωσης και πάχυνσης του αρθρικού υμένα. Ακόμα, έχουν επιρρέπεια για την ανάπτυξη βλαισογωνίας και ραιβογωνίας.
Συχνά οι κεφαλές των μηριαίων είναι υποπλαστικές και τα ισχία, βλαισά. Η προσβολή των μηριαίων κεφαλών οδηγεί σε προοδευτική και αναπηρική παραμόρφωση των ισχίων. Οι κλείδες είναι κοντές, παχειές και ακανόνιστες. Η λεκάνη, πτωχά σχηματισμένη. Τα μακρά οστά είναι κοντά, και οι διαφύσεις, πλατειές.
Πολλαπλή δυσόστωση παρουσιάζουν όλοι οι ασθενείς με βαριά MPS I. Τα νεογέννητα μπορεί να παρουσιάζουν ακτινολογικά ήπια δυσόστωση, ιδιαίτερα των ισχίων, όπως και πάχυνση των πλευρών (Clarke LA, 1997; Neufeld E and Muenzer J, 2001).
Οι ανωμαλίες των κέντρων οστεοποίησης των σπονδυλικών σωμάτων οδηγούν σε επιπέδωση και σχηματισμό ραμφοειδών προεξοχών των σπονδύλων και, συνακόλουθα, σε παραμορφώσεις της ΣΣ. Σπονδυλικές επιπλοκές περιλαμβάνουν παγίδευση σπονδυλικών νεύρων, οξεία σπονδυλική βλάβη και αστάθεια της ατλαντοαξονικής άρθρωσης. Θωρακο-οσφυική κύφωση συχνά γίνεται κλινικά εμφανής στη διάρκεια των 14 πρώτων μηνών της ζωής (Mundada and D’Souza, 2009).
Σύνδρομο καρπιαίου σωλήνα είναι συχνό σε όλες τις MPS, αλλά συχνά διαφεύγει της προσοχής, δοθέντος ότι εισβάλλει αθόρυβα και συνήθως παρουσιάζεται, εκτός από ατροφία του θέναρος, με λίγα συμπτώματα ή σημεία. Σε συνδυασμό με προσβολή των μεσοφαλαγγικών αρθρώσεων μπορεί να εξασθενήσει την λειτουργικότητα των χεριών (Clarke LA, 1997; Neufeld E and Muenzer J, 2001). Οφείλεται πιθανώς σε συνδυασμό αυξημένης λυσοσωμικής εναπόθεσης στον συνδετικό ιστό του καθεκτικού συνδέσμου και σε παραμορφώσεις συνεπεία των υποκείμενων οστικών δυσπλασιών.
Νευρολογικές διαταραχές.
Οι ασθενείς με βαριά MPS I, σε αντίθεση με τους ασθενείς με ήπια MPS I, αναπτύσσουν προοδευτική, βαριά διανοητική αναπηρία.
Η ανάπτυξή τους μπορεί αρχικά να είναι φυσιολογική, αλλά αργότερα, κατά τον 12ο μήνα της ηλικίας, αρχίζει προοδευτικά να εξασθενεί (Clarke LA, 1997) και, κατά τον 18ο μήνα της ηλικίας, αναπτύσσουν καθυστέρηση της διανοητικής ανάπτυξης.
Η καθυστέρηση της ανάπτυξης συνήθως σταθεροποιείται για μερικά χρόνια και ύστερα, βαθμιαία, οι διανοητικές ικανότητες περιορίζονται. Κατά το 8ο-10ο έτος της ηλικίας, οπότε οι ασθενείς με σύνδρομο Hurler καταλήγουν κακώς, οι περισσότεροι έχουν βαριά διανοητική αναπηρία.
Τα παιδιά με βαριά MPS I αναπτύσσουν περιορισμένες γλωσσικές ικανότητες, σχετιζόμενες πιθανώς με την αναπτυξιακή καθυστέρηση, την χρόνια απώλεια της ακοής και την διόγκωση της γλώσσας (Neufeld E and Muenzer J, 2001). Σε αντίθεση με την MPS II και την MPS III, οι βαριές αναπτυξιακές διαταραχές στα παιδιά με MPS I συνδέονται μάλλον με ήρεμη, παρά επιθετική, συμπεριφορά.
Η θειική ηπαράνη ανευρίσκεται σε αφθονία στον εγκέφαλο σαν μέρος της εξωκυττάριας θεμέλιας ουσίας. Η ανεπάρκεια της α-L-ιδουρονιδάσης σε ασθενείς με MPS I οδηγεί σε άθροιση γλυκοζαμινογλυκανών στα λυσοσώματα των νευρώνων και δευτεροπαθή άθροιση γλυκολιπιδίων, τα οποία σχηματίζουν σωμάτια τύπου «ζέβρας». Η εναπόθεση των GAG και των γλυκολιπιδίων στον εγκέφαλο ευθύνεται πιθανώς για την σοβαρή διανοητική αναπηρία και τον υδροκέφαλο.
Σπασμοί είναι ασυνήθιστοι, ακόμα και στα τελικά στάδια της νόσου (Clarke LA, 1997). Επικοινωνών υδροκέφαλος υψηλής πίεσης είναι συχνός σε ασθενείς με βαριά MPSI. Η ελαττωμένη απορρόφηση του ΕΝΥ αυξάνει την ενδοκρανιακή πίεση, οδηγώντας σε συμπίεση του εγκεφάλου. Σε μερικούς ασθενείς, η απότομη αύξηση της ενδοκρανιακής πίεσης μπορεί να προκαλέσει ραγδαία εξασθένηση της αναγνώρισης.
Οφθαλμικές ανωμαλίες.
Η όραση συχνά εξασθενεί λόγω θόλωσης του κερατοειδούς, η οποία εξελίσσεται προοδευτικά και μπορεί να οδηγήσει σε τύφλωση.
Οι θολερότητες του κερατοειδούς εμφανίζονται πρώιμα και αρχικά είναι περιφερικές, αλλά, κατά την 3η ή 4η δεκαετία της ζωής η δυστροφία του κερατοειδούς μπορεί να απειλήσει σοβαρά την όραση.
Γλαύκωμα ανοιχτής γωνίας μπορεί να παρατηρηθεί σε ασθενείς με βαριά MPS I (Clarke LA, 1997; Neufeld E and Muenzer J, 2001).
Εκφύλιση αμφιβληστροειδούς είναι συχνή και μπορεί να οδηγήσει σε εξασθένηση της περιφερικής όρασης και νυχτερινή τύφλωση. Η τύφλωση είναι αποτέλεσμα εκφύλισης του αμφιβληστροειδούς, συμπίεσης και ατροφίας του οπτικού νεύρου και βλάβης του φλοιού.
Ωτορινολαρυγγολογικές επιπλοκές.
Απώλεια της ακοής είναι συχνή σε ασθενείς με MPS I και σχετίζεται με την βαρύτητα της σωματικής νόσου. Στη γένεσή της συμβάλλουν κυρίως οι συχνές λοιμώξεις του μέσου ωτός λόγω δυσλειτουργίας των ευσταχιανών σαλπίγγων συνεπείας εναπόθεσης γλυκοζαμινογλυκανών μέσα στον στοματοφάρυγγα, δυσόστωσης των οσταρίων του μέσου ωτός, ουλοποίησης του τυμπάνου και βλάβης του 8ου νεύρου (Clarke LA, 1997; Neufeld E and Muenzer J, 2001).
Συχνά επίσης παρουσιάζουν χρόνια υποτροπιάζουσα ρινίτιδα με επίμονη, άφθονη ρινόρροια, χωρίς εμφανή λοίμωξη.
Αναπνευστικό.
Οι πάσχοντες από σύνδρομο Hurler συχνά αναπτύσσουν λοιμώξεις του ανώτερου και κατώτερου αναπνευστικού και διαταραχές της FVC.
Οι επιπλοκές από τις ανώτερες αναπνευστικές οδούς (Clarke LA, 1997; Neufeld EF and Muenzer J, 2001) οφείλονται στην άθροιση γλυκοζαμινογλυκανών στον στοματοφάρυγγα, σε συνδυασμό με υπερτροφία των αμυγδαλών και των αδενοειδών εκβλαστήσεων, στένωση της τραχείας και υπερεπιγλωττιδική στένωση, πάχυνση των φωνητικών χορδών, ανάπτυξη άφθονου ιστού στις ανώτερες αναπνευστικές οδούς και διόγκωση της γλώσσας (Peters ME et al, 1985; Shapiro J et al, 1985; Myer CM 3rd, 1991).
Οι επιπλοκές αυτές οδηγούν σε θορυβώδη αναπνοή, ιδιαίτερα στη διάρκεια της νύχτας, και είναι το συχνότερο αίτιο αποφρακτικής άπνοιας του ύπνου, η οποία αποτελεί συχνή επιπλοκή της νόσου, ιδιαίτερα στα όψιμα στάδια. Η προσβολή του ΚΝΣ μπορεί επίσης να συμβάλλει στην άπνοια του ύπνου. Οι ασθενείς με σύνδρομο Hurler μπορεί να έχουν βαθιά φωνή.
ΑΚΤΙΝΟΛΟΓΙΚΑ ΕΥΡΗΜΑΤΑ
Τα βαρέως πάσχοντα παιδιά με σύνδρομο Hurler παρουσιάζουν πρώιμα σοβαρές οστικές αλλοιώσεις. Στη γέννηση, ακτινολογικά μπορεί να παρουσιάζουν ήπια δυσόστωση, ιδιαίτερα του ισχίου, και πάχυνση των πλευρών (Clarke LA, 1997; Neufeld EF and Muenzer J, 2001).
Προοδευτική σκελετική δυσπλασία (πολλαπλή δυσόστωση), η οποία περιλαμβάνει όλα τα οστά, αναπτύσσουν όλοι οι ασθενείς με σύνδρομο Hurler.
Θώρακας-πλευρές
- Βράχυνση, πάχυνση και ακανόνιστου σχήματος κλείδες και υποπλασία των ωμοπλατών
- Πλευρές σχήματος «κουπιού» (στενές στα σπονδυλικά και πλατειές, στα στερνικά άκρα)
Κρανίο
- Διόγκωση τουρκικού εφιππίου σχήματος «J»
- Κρανιοσυνοστέωση
- Μακροκεφαλία
- Πάχυνση των οστών του κρανίου
- Πρόωρη σύγκλειση των οβελιαίων και λαβδοειδών ραφών
Λεκάνη-ισχία
- Αρθροκατάδυση (ενίοτε)
- Γωνίωση ιεροϊσχιακής εντομής
- Στένωση αυχένα μηριαίων
- Υποπλασία και στένωση των βάσεων των λαγονίων με ψευδο-διόγκωση της κοτύλης και βλαισότητα του ισχίου
- Υποπλασία των μηριαίων κεφαλών, η οποία οδηγεί σε προοδευτική και αναπηρική παραμόρφωση του ισχίου
Σπονδυλική στήλη
- Δυσπλασία των σπονδυλικών σωμά των με αμφίκυρτες τελικές πλάκες και ωοειδή, αγκιστροειδούς σχήματος, διαμόρφωση του σώματος των κατώτερων θωρακικών και ανώτερων οσφυικών σπονδύλων
- Πρόσθια υποπλασία των οσφυικών σπονδύλων με κύφωση (πρώιμο εύρημα)
- Στένωση του αυχένα των σπονδυλικών τόξων
- Υποπλασία οδοντοειδούς απόφυσης
- Υπεξάρθρημα ατλαντοαξονικής
- Στυλοϋοειδής σύνδεσμος
- Σχεδόν πάντα ο στυλοϋοειδής σύνδεσμος ασβεστοποιείται και γίνεται παχύτερος από το φυσιολογικό
Χέρια
- Βράχυνση και αποπλάτυνση των φαλάγγων οι οποίες αποκτούν σχήμα «βλήματος», με όξυνση του εγγύς τμήματος του 2ου – 5ου μετακαρπίου
Αλλα ευρήματα
- Βράχυνση και διαπλάτυνση των διαφύσεων των μακρών σωληνωδών οστών με μικρές, παραμορφωμένες επιφύσεις
- Λέπτυνση του φλοιού των οστών
- Οστεοπόρωση
1.2. ΗΠΙΑ ΒΛΕΝΝΟΠΟΛΥΣΑΚΚΧΑΡΙΔΩΣΗ ΤΥΠΟΥ Ι
ΣΥΝΩΝΥΜΑ
- Ηπια MPS I (attenuated MPS I)
- Σύνδρομο Hurler-Scheie/σύνδρομο Scheie (Hurler-Scheie syndrome/Scheie syndrome) (πρώην MPS V)
ΠΕΡΙΓΡΑΦΗ ΝΟΣΟΥ
Οι ασθενείς με MPS I που έχουν φυσιολογική ανάπτυξη και μέτριες σωματικές ανωμαλίες κατά τον 24ο μήνα της ηλικίας, θεωρούνται ότι έχουν ήπια (εξασθενημένη) MPS I. Η ήπια MPS I παρουσιάζεται συνήθως μεταξύ 3ου-10ου έτους της ηλικίας. Οι ασθενείς με ήπια MPS I μπορεί να έχουν φυσιολογική ψυχοκινητική ανάπτυξη στην πρώιμη παιδική ηλικία, αλλά και μαθησιακές δυσκολίες.
Η βαρύτητα και ο βαθμός της εξέλιξης της νόσου κυμαίνεται από σοβαρές, απειλητικές για την ζωή, επιπλοκές οι οποίες οδηγούν στο θάνατο την 2η έως 3η δεκαετία της ζωής, έως φυσιολογική διάρκεια ζωής επιπλεκόμενη από σοβαρή αναπηρία λόγω προοδευτικών αρθρικών εκδηλώσεων. Απώλεια της ακοής και βαλβιδοπάθεια της καρδιάς είναι συχνή σε ασθενείς με MPS I.
ΚΛΙΝΙΚΕΣ ΕΚΔΗΛΩΣΕΙΣ ΗΠΙΑΣ MPS I
Αναπνευστικές.
Συχνά, οι ασθενείς με ήπια MPS I παρουσιάζουν άπνοια ύπνου, λόγω απόφραξης των αεραγωγών και πιθανώς προσβολής του ΚΝΣ, και προοδευτική πνευμονοπάθεια με διαταραχές της FVC. Οι αναπνευστικές επιπλοκές (όπως και οι καρδιαγγειακές) είναι μία από τις κύριες αιτίες πρόωρου θανάτου.
Γαστρεντερικές.
Ηπατομεγαλία, διάφορου βαθμού, είναι συχνή σε ασθενείς με ήπια MPS I. Κήλες παρουσιάζει το 65% περίπου των ασθενών με ήπια MPS I (Thomas JA et al, 2010), λιγότερο συχνά από τους ασθενείς με βαριά MPS I.
Καρδιαγγειακές.
Η καρδιά προσβάλλεται στο 88% περίπου των παιδιών με ήπια MPS I και σε ηλικία 11.7, κατά μέσον όρο, ετών (Thomas JA et al, 2010). Η προσβολή της καρδιάς παρουσιάζεται με παλινδρόμηση ή/και στένωση της μιτροειδούς και της αορτικής βαλβίδας, η οποία μπορεί να χρειασθεί χειρουργική αντικατάσταση.
Η βαλβιδοπάθεια της αορτής είναι συχνότερη σε ασθενείς με ήπια, παρά με βαριά, MPS Ι (Neufeld EF and Muenzer J, 2001), αν και, σε μερικούς ασθενείς, προσβάλλονται όλες οι βαλβίδες. Η στεφανιαία νόσος μπορεί ακόμα να είναι εκδήλωση της ήπιας MPS I.
Κρανιοπροσωπικές ανωμαλίες.
Η τράχυνση των χαρακτηριστικών του προσώπου είναι λιγότερο εμφανής σε ασθενείς με ήπια, παρά με βαριά, MPS I. Οι ασθενείς με ήπια MPS I έχουν βραχύ αυχένα, μεγάλο στόμα και τατράγωνη και μικρή γνάθο και αναστολή της ανάπτυξης.
Νευρολογικές.
Οι ασθενείς με ήπια MPS I έχουν φυσιολογική ή σχεδόν φυσιολογική διανόηση. Η εξασθένηση των διανοητικών ικανοτήτων έχει περισσότερο παρατεταμένη διαδρομή στην ήπια απ΄ό,τι στη βαριά MPS I. Όπως και στη βαριά MPS I, υδροκέφαλος αναπτύσσεται και στην ήπια MPS I, αλλά λιγότερο συχνά, όπως και αραχνοειδείς κύστεις.
Προοδευτική συμπίεση του νωτιαίου μυελού, η οποία καταλήγει σε αυχενική μυελοπάθεια και οφείλεται σε πάχυνση της σκληρής μήνιγγας είναι συχνή σε ασθενείς με ήπια MPS I. Η αυχενική μυελοπάθεια μπορεί να παρουσιασθεί αρχικά σαν μειωμένη δραστηριότητα ή δυσανεξία στις ασκήσεις, και να διαπιστωθεί μόνον όταν η βλάβη είναι μόνιμη και μη αναστρέψιμη (Neufeld EF and Muenzer J, 2001).
Οφθαλμολογικές.
Θολερότητες του κερατοειδούς αναπτύσσει το 82% περίπου των παιδιών με ήπια MPS I κατά το 9ο, κατά μέσον όρο, έτος της ηλικίας (Thomas JA et al, 2010) και μπορεί να οδηγήσουν σε σοβαρή εξασθένηση της όρασης. Γλαύκωμα, εκφύλιση του αμφβληστροειδούς και ατροφία του οπτικού νεύρου μπορεί επίσης να παρατηρηθούν.
Σκελετικές.
Οι σκελετικές και αρθρικές εκδηλώσεις είναι οι κυριότερες αιτίες αναπηρίας και ενοχλημάτων σε ασθενείς με ήπια MPS I (Clarke LA, 1997; Vijay S and Wraith JE, 2005). Οι ασθενείς με ήπια MPS I μπορεί να έχουν σοβαρές σκελετικές ανωμαλίες, χωρίς εξασθένηση της αναγνώρισης.
Συχνά έχουν κύφωση, σκολίωση και σοβαρή οσφυαλγία, όπως και σπονδυλολίσθηση της κατώτερης ΣΣ, η οποία οδηγεί σε συμπίεση του νωτιαίου μυελού, σε ασθενείς με ενδιάμεση νόσο.
Περισσότεροι από 85% των ασθενών με ήπια MPS I έχουν δυσόστωση, κυρίως στην ΣΣ και τα μηριαία (Thomas JA et al, 2010). Ολοι οι ασθενείς με ήπια MPS I αναπτύσσουν προοδευτική προσβολή όλων των αρθρώσεων, η οποία οδηγεί σε απώλεια ή σοβαρό περιορισμό του εύρους της κίνησης.
Συχνά ακόμα έχουν πτωχή λειτουργικότητα των χεριών σαν αποτέλεσμα γαμψοδακτυλίας, συνδρόμου καρπιαίου σωλήνα και δυσκαμψίας των μεσοφαλαγγικών αρθρώσεων. Σύνδρομο καρπιαίου σωλήνα παρουσιάζει το 67% των ασθενών με ήπια MPS I, κατά το 13ο, κατά μέσον όρο, έτος της ηλικίας (Thomas JA et al, 2010).
Πάνω από 90% των ασθενών με ήπια MPS I έχουν κοιλοποδία, βλαισογωνία και βαδίζουν στα ακροδάκτυλα (Thomas JA et al, 2010).
Ωτορινολαρυγγολογικές.
Μέτρια έως σοβαρή απώλεια της ακοής αναπτύσσουν πολλοί ασθενείς με ήπια MPS I, ιδιαίτερα τα παιδιά με σοβαρές σωματικές ανωμαλίες. Η βαρηκοΐα, συνήθως στις υψηλές συχνότητες, οφείλεται μάλλον σε δυσλειτουργία των ευσταχιανών σαλπίγγων, δυσόστωση των οσταρίων του μέσου ωτός και προσβολή του 8ου νεύρου. Ρινόρροια είναι συχνή.
ΚΛΙΝΙΚΕΣ ΕΚΔΗΛΩΣΕΙΣ MPS I (ΣΥΝΟΠΤΙΚΑ)
Αισθητήρια όργανα
- Βαρηκοΐα
- Γλαύκωμα ανοιχτής γωνίας
- Διόγκωση της κεφαλής του οπτικού νεύρου
- Εκφυλιστική αμφιβληστροειδοπάθεια
- Θόλωση του κερατοειδούς (συχνά)
Αναπνευστικές επιπλοκές
- Αναπνευστικές λοιμώξεις
- Διαταραχές της FVC
- Ρινίτιδα
- Βαθιά φωνή
Γαστρεντερικές διαταραχές
- Απώλεια κοπράνων και διάρροιες, εναλλασσόμενες ενίοτε με σοβαρή δυσκοιλιό-τητα
- Βουβωνοκήλες/ομφαλοκήλες
- Προβολή της κοιλιάς λόγω προοδευτικής ηπατοσπληνομεγαλίας
Δέρμα και εξαρτήματα
- Μελανοκυττάρωση
- Ξηρότητα, ωχρότητα και τράχυνση του δέρματος
- Υπερτρίχωση σώματος και προσώπου
Καρδιαγγειακό σύστημα
- Αιφνίδιος θάνατος από αρρυθμία
- Ενδοκαρδιακή ινοελάστωση
- Καρδιαγγειακό collapsus
- Μυοκαρδιοπάθεια
- Παλινδρόμηση μιτροειδούς και αορτής
- Στεφανιαία νόσος
Κρανιοπροσωπικές ανωμαλίες
- Βράχυνση του αυχένα
- Μακροκεφαλία με μετωπιαίο ύβο
- Σκαφοειδοκεφαλία (συχνά)
- Κρανιομεγαλία, με πάχυνση του θόλου του κρανίου και αβαθείς οφθαλμικοί κόγχοι
- Τράχυνση των χαρακτηριστικών του προσώπου (γκαργκοϋλικό προσωπείο)
- Πάχυνση των παρειών
- Προέχουσες ρινοχειλικές πτυχές
- Προέχοντα κάτω βλέφαρα
- Πάχυνση των λοβών των πτερυγίων των ώτων
- Συμπίεση της ρινικής γέφυρας, με αποπλάτυνση της κορυφής της ρινός και αναστροφή των μυκτήρων
- Συνοφρυδία
- Υπερτρίχωση προσώπου
Μυοσκελετικές ανωμαλίες
- Αναστολή ανάπτυξης
- Ατροφία των μυών του θέναρος
- Βλαισογωνία/ραιβογωνία
- Βράχυνση, πάχυνση και ακανόνιστη διαμόρφωση κλειδών
- Βράχυνση μακρών οστών και διαπλάτυνση των μεταφύσεων
- Δυσκαμψία/συγκάμψεις αρθρώσεων-δυσκαμψία θωρακικού τοιχώματος
- Σπονδυλικές ανωμαλίες :
- Θωρακική σκολίωση με/ή χωρίς οσφυική κύφωση
- Επιπέδωση και σχηματισμός ραμφοειδών προεξοχών των σπονδύλων
- Παγίδευση σπονδυλικών νεύρων
- Αστάθεια ατλαντοαξονικής άρθρωσης
- Υποπλασία οδοντοειδούς απόφυσης
- Σύνδρομο καρπιαίου σωλήνα
- Τροπιδοειδής θώρακας
- Κωνοειδής θώρακας
- Υποπλασία λεκάνης
- Υποπλασία μηριαίας κεφαλής/βλαισότητα ισχίων
Νευρολογικές ανωμαλίες
- Διανοητική καθυστέρηση
- Ελάττωση αντανακλαστικών κατάποσης
- Επικοινωνών υδροκέφαλος υψηλής πίεσης
- Καθυστέρηση μυελινοποίησης, ατροφία και διεύρυνση των κοιλιών
- Σπασμοί
- Σύνδρομο καρπιαίου σωλήνα
- Υπαραχνοειδείς κύστεις
- Ψυχωσικά επεισόδια
Ανωμαλίες στόματος
- Ανοιχτό συνήθως στόμα, ιδιαίτερα μετά το 3ο έτος της ηλικίας
- Διόγκωση της γλώσσας και των χειλέων
- Μικροί και αραιοί οδόντες
ΔΙΑΓΝΩΣΗ MPS I
Η MPS I πρέπει να μπαίνει στη σκέψη σε ασθενείς με τις εξής εκδηλώσεις :
- Τράχυνση των χαρακτηριστικών του προσώπου
- Ηπατοσπληνομεγαλία
- Χαρακτηριστικές σκελετικές και αρθρικές εκδηλώσεις (ύβος, περιορισμός κινη-τικότητας αρθρώσεων)
- Χαρακτηριστικές οφθαλμικές ανωμαλίες (θολερότητες κερατοειδούς)
Η διάγνωση της MPS I βασίζεται στην ανεύρεση της ανεπάρκειας της α-L-ιδουρονιδάσης σε περιφερικά λευκά αιμοσφαίρια, στο πλάσμα ή σε καλλιεργημένους ινοβλάστες. Η αυξημένη αποβολή θειικής ηπαράνης και δερματάνης από τα ούρα βοηθά στη διάγνωση.
Η προνεογνική διάγνωση μπορεί να γίνει με την αναζήτηση του ενζύμου σε καλλιεργημένα κύτταρα αμνιακού υγρού ή σε χοριακές λάχνες. Εάν η οικογένεια των πασχόντων έχει γνωστή ειδική μετάλλαξη, η ανάλυση του DNA είναι περισσότερο αξιόπιστη για την προνεογνική διάγνωση της MPS I.
ΔΙΑΦΟΡΙΚΗ ΔΙΑΓΝΩΣΗ MPS I
- Α-μαννοσίδωση
- Βλεννολιπίδωση (σιαλίδωση τύπου ΙΙ)
- Βλεννολιπίδωση τύπου ΙΙ (νόσος των Ι-κυττάρων)
- Βλεννολιπίδωση ΙΙΙ α/β
- Βλεννοπολυσακκχαρίδωση τύπου II
- Βλεννοπολυσακκχαρίδωση τύπου III
- Βλεννοπολυσακκχαρίδωση τύπου IV
- Βλεννοπολυσακκχαρίδωση τύπου VI
- Βλεννοπολυσακκχαρίδωση τύπου VII
- Νεανική ιδιοπαθής αρθρίτιδα
- Πολλαπλή ανεπάρκεια σουλφατάσης
Οι εκδηλώσεις της MPS I επικαλύπτουν τις εκδηλώσεις άλλων λυσοσωμικών νοσημάτων, ιδιαίτερα των βλεννοπολυσακκχαριδώσεων, όπως η πολλαπλή ανεπάρκεια σουλφατάσης, η βλεννολιπίδωση (σιαλίδωση τύπου ΙΙ), η βλεννολιπίδωση τύπου ΙΙ (νόσος των Ι-κυττάρων), η βλεννολιπίδωση ΙΙΙ α/β και η α-μαννοσίδωση. Η διάκριση γίνεται με βάση τα κλινικά ευρήματα και τις βιοχημικές αναλύσεις.
Πρέπει να σημειωθεί ότι ανεπάρκεια της α-L-ιδουρονιδάσης μπορεί να παρατηρηθεί και στη νόσο των Ι-κυττάρων (βλεννολιπίδωση ΙΙ) και στην πολυδυστροφία ψευδο-Hurler (βλεννολιπίδωση III α/ β). Στα νοσήματα αυτά, η α-L-ιδουρονιδάση συντίθεται σε επαρκείς ποσότητες, αλλά δεν μεταφέρεται στα λυσοσώματα λόγω διαταραχής της διαδικασίας στόχευσης των λυσοσωμάτων της επαγόμενης μέσω υποδοχέων (receptor-mediated lysosomal targeting). Ακόμα, οι ασθενείς με πολυδυστροφία ψευδο-Hurler έχουν φυσιολογική απέκκριση GAG, καθαρούς κερατοειδείς και δεν πάσχουν από διανοητική καθυστέρηση.
Οι ασθενείς με ήπια MPS I μπορεί να παρουσιασθούν με μη φλεγμονώδη αρθρίτιδα σε οποιαδήποτε ηλικία, γι΄αυτό και η MPS I πρέπει να μπαίνει στη ΔΔ της νεανικής ιδιοπαθούς αρθρίτιδας (Cimaz R et al, 2006).
Η προσεκτική κλινική εξέταση για άλλες εκδηλώσεις MPS I, όπως και οι χαρακτήρες της αρθρικής προσβολής, επιτρέπει την διάκριση των ασθενών με ήπια MPS I που παρουσιάζονται με μη φλεγμονώδη αρθρίτιδα από την ΝΙΑ.
ΘΕΡΑΠΕΙΑ
1. ΘΕΡΑΠΕΙΑ ΕΚΔΗΛΩΣΕΩΝ - ΕΠΙΠΛΟΚΩΝ
Απνοια ύπνου
- Τραχειοτομία ή αερισμός συνεχούς θετικής πίεσης με συμπληρωματικό οξυγόνο.
- Τραχειοστομία, για να διατηρηθεί η βατότητα των αναπνευστικών οδών και να τεθεί υπό έλεγχο η άπνοια ύπνου, η πνευμονική υπέρταση και η δεξιά καρδιακή ανεπάρκεια
Απώλεια ακοής
- Αμυγδαλεκτομή και αδενοειδεκτομή, για την δυσλειτουργία των ευσταχιανών σαλπίγγων ή/και την απόφραξη των ανώτερων αναπνευστικών οδών
- Βοηθήματα ακοής
- Τοποθέτηση αεραγωγών (σε βαρέως πάσχοντες ασθενείς)
Γαστρεντερικές διαταραχές
- Η διάρροια και η δυσκοιλιότητα αντιμετωπίζονται συμπτωματικά με δίαιτα και η δυσκοιλιότητα, με καθαρκτικά.
Καρδιαγγειακές ανωμαλίες
- Αντικατάσταση καρδιακών βαλβίδων (εάν χρειάζεται)
Κήλες
- Οι βουβωνοκήλες αντιμετωπίζονται χειρουργικά.
- Οι ομφαλοκήλες δεν χρειάζονται θεραπεία, εκτός εάν αποκτήσουν μεγάλο μέγεθος και δημιουργούν προβλήματα.
- Μυοσκελετικές ανωμαλίες
Φυσιοθεραπεία.
Παίζει σημαντικό ρόλο στη θεραπεία της MPS I (Tylki- Szymanska A et al, 2010a). Οι ασκήσεις εύρους κίνησης, εάν αρχίσουν πρώιμα, μπορεί να διατηρήσουν την λειτουργικότητα των αρθρώσεων. Εάν οι αρθρώσεις έχουν μεγάλο περιορισμό της κινητικότητας, το εύρος τους αυξάνεται μόνο μετά από HSCT
ΧΕΙΡΟΥΡΓΙΚΗ ΘΕΡΑΠΕΙΑ
- Διορθωτικές χειρουργικές επεμβάσεις, για τις συγκάμψεις των αρθρώσεων και παραμορφώσεων των χεριών και των ποδιών
- Ολικές αρθροπλαστικές
- Σταθεροποίηση ατλαντοϊνιακής άρθρωσης
- Η αυχενική μυελοπάθεια η οφειλόμενη σε συμπίεση του νωτιαίου μυελού πρέπει να εκτιμάται και να αντιμετωπίζεται έγκαιρα
Οφθαλμικές ανωμαλίες
- Οφθαλμικά σκίαστρα, για να περιορισθεί το θάμβος της όρασης το οφειλόμενο στις θολερότητες του κερατοειδούς
- Μεταμόσχευση κερατοειδούς σε ασθενείς με ήπια νόσο, εάν οι θολερότητες του κερατοειδούς εξασθενούν σημαντικά την όραση, αν και τα μοσχεύματα στη συνέχεια θολώνουν. Ακόμα όμως και αν τα μοσχεύματα είναι διαυγή, οι πάσχοντες μπορεί να συνεχίσουν να έχουν εξασθένηση της όρασης λόγω προσβολής του αμφιβληστροειδούς ή/και του οπτικού νεύρου (Neufeld E and Muenzer J, 2001).
Σύνδρομο καρπιαίου σωλήνα
- Χειρουργική αποσυμπίεση του μέσου νεύρου. Συνιστάται ιδιαίτερα σε ασθενείς με ήπια MPS I και σε πάσχοντες από σοβαρή MPS I που έχουν θεραπευθεί με HSCT. Στους περισσότερους ασθενείς με MPS I τα τυπικά συμπτώματα τα συμπίεσης του μέσου νεύρου (πόνος, «γαργάλημα», αιμωδία) απουσιάζουν μέχρις ότου η συμπίεση γίνει σοβαρή (Haddad FS et al, 1997; Van Heest AE et al, 1998; Bahadir C et al, 2009). Γι΄αυτό και πρέπει να γίνονται μελέτες της αγωγιμότητας στα αρχικά στάδια της νόσου για να να εντοπισθούν οι ασθενείς με σύνδρομο καρπιαίου σωλήνα πρόωρα, οπότε η χειρουργική επέμβαση έχει καλύτερο αποτέλεσμα. Η χειρουργική αποσυμπίεση του μέσου νεύρου σε ασθενείς με σύνδρομο καρπιαίου σωλήνα έχει ποικίλα αποτελέσματα στην αποκατάσταση της κινητικότητας των χεριών (Van Heest AE et al, 1998). Μερικοί ασθενείς χρειάζονται επανειλημμένες χειρουργικές επεμβάσεις (Neufeld E and Muenzer J, 2001).
Υδροκέφαλος : Κοιλιοπεριτοναϊκή παροχέτευση
2. ΠΡΟΛΗΨΗ ΠΡΩΤΟΠΑΘΩΝ ΕΚΔΗΛΩΣΕΩΝ
2.1 ΕΝΖΥΜΙΚΗ ΘΕΡΑΠΕΙΑ ΑΝΑΠΛΗΡΩΣΗΣ (Enzyme Replacement Therapy; ERT)
Η λαρονιδάση (Aldurazyme) είναι μία πολυμορφική ποικιλία του ανθρώπινου ενζύμου α-L-ιδουρονιδάση, παραγόμενη με τεχνολογία ανασυνδυασμένου DNA.
Ενδείξεις :
- Θεραπεία μη νευρολογικών εκδηλώσεων MPS I
- Αντενδείξεις/άρνηση ασθενών για θεραπεία με BMT
Η ERT μπορεί να προηγηθεί της μεταμόσχευσης του μυελού των οστών (HSCT) για να μειωθεί το φορτίο της εναπόθεσης και να βελτιωθούν οι πνευμονικές και καρδιακές εκδηλώσεις, ώστε να μετριασθούν οι μεταμοσχευτικές επιπλοκές (Tolar J et al, 2008).
Αποτελεσματικότητα
Η αποτελεσματικότητα της ERT εξαρτάται από την ικανότητα των ενδοφλέβια χορηγούμενων ανασυνδυασμένων ενζύμων να εισδύσουν στα κύτταρα των διάφορων ιστών και να αθροισθούν στα λυσοσώματα (Russell CS and Clarke LA, 1999).
- Βελτιώνει την ανάπτυξη (Kakkis ED et al 2001; Sifuentes M et al 2007; Clarke LA et al, 2009).
- Βελτιώνει την κινητικότητα των αρθρώσεων (Kakkis ED et al 2001; Sifuentes M et al, 2007; Clarke LA et al, 2009)
- Αυξάνει την ικανότητα εκτέλεσης των καθημερινών δραστηριοτήτων και (Kakkis ED et al, 2001; Sifuentes M et al, 2007).
- Σταθεροποιεί την προσβολή της καρδιάς, αλλά δεν καθυστερεί την εξέλιξη της προσβολής των καρδιακών βαλβίδων (Kakkis ED et al, 2001; Sifuentes M et al, 2007).
- Βελτιώνει την άπνοια του ύπνου, την αναπνοή και την πνευμονική λειτουργία (Kakkis ED et al 2001; Wraith EJ et al, 2005; Sifuentes M et al, 2007; Clarke LA et al, 2009)
- Ελαττώνει τον όγκο του ήπατος (Clarke LA et al, 2009; Kakkis ED et al, 2001; Wraith EJ et al, 2005; Sifuentes M et al, 2007).
- Μειώνει ή φυσιολογικοποιεί τα επίπεδα των απεκκρινόμενων από τα ούρα GAG (Wraith EJ et al, 2005; Clarke LA et al, 2009)
- Βελτιώνει την ποιότητα της ζωής, ιδιαίτερα όσον αφορά τον πόνο (Clarke LA et al, 2009).
- Βελτιώνει την οπτική οξύτητα, αν και δεν επηρεάζει τις θολερότητες του κερατοειδούς (Clarke LA et al, 2009).
- Βελτιώνει το ύψος και την διάμετρο του κρανίου, εάν χορηγηθεί πρώιμα (κάτω από το 1ο έτος της ηλικίας) (Tylki-Szymanska A et al, 2010b)
- Μπορεί να βελτιώσει την ικανότητα μάθησης (Valayannopoulos V et al, 2010).
Σε βαρέως πάσχοντες η λαρονιδάση δεν αποτρέπει την εξασθένηση της αναγνώρισης, δεδομένου ότι, στις συνιστώμενες δόσεις (100 IU/kg/εβδομάδα) δεν διέρχεται μέσω του αιματοεγκεφαλικού φραγμού σε σημαντικές ποσότητες. Χορηγούμενη ενδοθηκικά σε ασθενείς με προσβολή του νωτιαίου μυελού, μειώνει τα επίπεδα των GAG στο ΕΝΥ και την πίεση του ΕΝΥ (Munoz -Rojas MV et al, 2008).
Δοσολογικό σχήμα
- Προθεραπευτική χορήγηση αντιφλεγμονωδών και αντιϊσταμινικών
- Ενδοφλέβια έγχυση Aldurazyme 100 U/kg/εβδομάδα σε διάστημα 4 ωρών
- Ενήλικες :
- 0.58 mg/kg IV qwk σε διάστημα 4 ωρών
- Ενδοφλέβια έγχυση με ρυθμό 10 mcg/kg/ώρα και βαθμιαία αύξηση q15 min όσο γίνεται ανεκτή μέσα στην 1η ώρα (όχι >200 mcg/kg/ώρα)
- Παιδιά : >5 ετών δόση ενηλίκων
2.2 ΜΕΤΑΜΟΣΧΕΥΣΗ ΑΙΜΟΠΟΙΗΤΙΚΩΝ ΒΛΑΣΤΙΚΩΝ ΚΥΤΤΑΡΩΝ [(Hema-topoietic Stem Cell Transplantation; HSCT)]Λ
Ενδείξεις :
- Θεραπεία μη νευρολογικών εκδηλώσεων MPS I. Η HSCT πρέπει να χρησιμοποιείται μόνο σε προσεκτικά επιλεγμένα παιδιά με προσεκτική και ενδελεχή προ-μεταμοσχευτική κλινική αξιολόγηση, στα οποία θα είναι δυνατή η μακροχρόνια συστηματική παρακολούθηση (Neufeld E and Muenzer J, 2001).
Αποτελεσματικότητα.
Η ωφέλιμη δράση της HSCT οφείλεται σε αντικατάσταση των ανεπαρκούντων μακροφάγων από μακροφάγα μυελού των οστών δωρητών (κύτταρα Kupffer, πνευμονικά, σπληνικά, αμυγδαλικά, περιτοναϊκά και οζώδη μακροφάγα και μικρογλοιακά κύτταρα) (Guffon N et al, 1998; Prasad VK and Kurtzberg J, 2010). Η αποτυχία της στη θεραπεία των σκελετικών εκδηλώσεων αποδίδεται στη σχετικά πτωχή αγγείωση του οστίτη ιστού (Taylor C et al, 2008).
Η αποτελεσματικότητα της HSCT εξαρτάται γενικά από τον βαθμό της κλινικής προσβολής και την ηλικία του παιδιού στον χρόνο της μεταμόσχευσης. Γενικά, η HSCT έχει καλύτερο αποτέλεσμα εάν γίνει πριν από το 2ο έτος της ηλικίας.
Η HSCT καθυστερεί την εξέλιξη μερικών εκδηλώσεων σε παιδιά με σοβαρή MPS I (Vellodi A et al, 1997; Guffon N et al, 1998; Neufeld E and Muenzer J, 2001; Souillet G et al, 2003; Staba SL et al, 2004) :
- Μειώνει την διόγκωση του ήπατος και του σπληνός και την τράχυνση των χαρακτηριστικών του προσώπου, βελτιώνει την ακοή και διατηρεί την καρδιακή λειτουργία σε φυσιολογικά επίπεδα
- Δεν επηρεάζει τις σκελετικές εκδηλώσεις και τις θολερότητες του κερατοειδούς, οι οποίες συνεχίζουν να εξελίσσονται στον ίδιο βαθμό όπως και στα μη θεραπευόμενα παιδιά (Weisstein JS et al, 2004, Taylor C et al, 2008)
- Δεν είναι γνωστό κατά πόσον βελτιώνει τις νευρολογικές επιπλοκές (εκτός από την προοδευτική εξασθένηση της διανόησης). Μερικοί υποστηρίζουν ότι έχει αποτέλεσμα στις επιπλοκές αυτές (Munoz-Rojas MV et al, 2008; Valayannopoulos V et al, 2010), άλλοι όμως όχι.
- Σε παιδιά θεραπευθέντα με HSCT πριν από την εμφάνιση σημαντικής αναπτυξιακής καθυστέρησης (δηλ. συνήθως μεταξύ 12ου-18ου μήνα της ηλικίας) φαίνεται ότι καθυστερεί την εξασθένηση της λειτουργίας της αναγνώρισης. Τα παιδιά με εγκατεστημένη σοβαρή εξασθένηση της αναγνώρισης δεν ανταποκρίνονται στην HSCT.
- Δεν θεραπεύει και δεν βελτιώνει την προσβολή των καρδιακών βαλβίδων ή τις σκελετικές εκδηλώσεις
- Σε ασθενείς με σοβαρή MPS I σταθεροποιεί και βελτιώνει την μυοκαρδιακή λειτουργία, με ύφεση της υπερτροφίας και επάνοδο των διαστάσεων των διαμερισμάτων της καρδιάς σε φυσιολογικά όρια, αν και δεν φαίνεται να βελτιώνει σημαντικά τις βαλβιδικές αλλοιώσεις (Braunlin EA et al, 2003).
- Aυξάνει την διάρκεια της επιβίωσης κατά 67%, συγκριτικά με τους μη θεραπευόμενους με HSCT (Moore D et al, 2008)
Επιπλοκές.
Η HSCT συνδέεται με σοβαρές καρδιακές και πνευμονικές επιπλοκές (Neufeld E and Muenzer J, 2001; Gassas A et al, 2003).
Συχνά ακόμα επιπλέκεται με αποτυχία σταθεροποίησης του μοσχεύματος και νόσο μοσχεύματος έναντι του ξενιστή (Peters et al, 1996; Peters et al, 1998a; Peters et al, 1998b), γι΄αυτό και συνοδεύεται από μεγάλη νοσηρότητα και θνητότητα (Neufeld E and Muenzer J, 2001). Τα αντισώματα έναντι της α-L-ιδουρονιδάσης δεν φαίνεται να ευθύνονται για την απόρριψη του μοσχεύματος (Soni S et al, 2007; Tolar J et al, 2008).
Οι ασθενείς που έχουν θεραπευθεί με HSCT, εν μέρει λόγω αύξησης του προσδόκιμου επιβίωσης, συχνά παρουσιάζουν αυξανόμενο πόνο και δυσκαμψία των ισχίων και των γονάτων, συμπίεση του νωτιαίου μυελού, σύνδρομο καρπιαίου σωλήνα και προοδευτική θωρακο-οσφυική κύφωση (Neufeld E and Muenzer J, 2001), γι΄ αυτό και, μετά την HSCT, συχνά χρειάζονται διάφορες ορθοπεδικές επεμβάσεις για να διατηρήσουν την λειτουργικότητά τους και την βάδιση (Masterson EL et al 1996; Tandon V et al 1996).
3. ΠΡΟΛΗΨΗ ΔΕΥΤΕΡΟΠΑΘΩΝ ΕΠΙΠΛΟΚΩΝ
- Προφύλαξη από βακτηριδιακή ενδοκαρδίτιδα, σε ασθενείς με καρδιακές ανωμαλίες (Neufeld E and Muenzer J, 2001)
- Σωστή τοποθέτηση και αποφυγή υπερέκτασης του αυχένα, λόγω της αστάθειας της ατλαντοϊνιακής
- Λόγω στένωσης της τραχείας και της πάχυνσης των φωνητικών χορδών, η διασωλήνωση μπορεί να χρειασθεί μικρότερους του κανονικού ενδοτραχειακούς σωλήνες ή ινοοπτικό λαρυγγοσκόπιο.
- Ειδική προσοχή στους κινδύνους της αναισθησίας, λόγω της δυσκολίας στη βατότητα των αεραγωγών. Γενικά, η γενική αναισθησία πρέπει να δίνεται σε Κέντρα επανδρωμένα με αναισθησιολόγους με εμπειρία στο χειρισμό των ασθενών με βλεννοπολυσακκχαριδώσεις (Neufeld E and Muenzer J, 2001)
.
2. ΒΛΕΝΝΟΠΟΛΥΣΑΚΧΑΡΙΔΩΣΗ ΤΥΠΟΥ II (σύνδρομο Hunter) (Mucopolysaccharidosis type II; MPS II; Hunter syndrome)
ΧΑΡΑΚΤΗΡΙΣΤΙΚΑ ΝΟΣΟΥ
Η MPS II (σύνδρομο Hunter; McKusick 309900) πρωτοπεριγράφηκε το 1917 από τον Hunter. Πρόκειται για Χ-φυλοσύνδετο νόσημα του μεταβολισμού των πολυσακκχαριδών, οφειλόμενο σε ανεπάρκεια της ιδουρονικής σουλφατάσης (iduronate sulfatase) και συνακόλουθη άθροιση ηπαράνης και θειικής δερματάνης σε διάφορους ιστούς και όργανα.
Κλινικά, χαρακτηρίζεται από:
- δυσόστωση με νανισμό
- γκροτέσκο προσωπείο,
- ηπατοσπληνομεγαλία,
- καρδιαγγειακές ανωμαλίες και
- κώφωση.
Το σύνδρομο Hunter ξεχωρίζει από τις άλλες MPSs επειδή, λόγω της Χ-φυλοσύνδετης μετάδοσής της, επικρατεί στους άρρενες. Ακόμα, θολερότητες του κερατοειδούς στο σύνδρομο Hunter απουσιάζουν.
ΕΠΙΔΗΜΙΟΛΟΓΙΑ
Συχνότητα. Η συχνότητα της MPS ΙΙ ποικίλλει ευρέως. Υπολογίζεται σε 1 περίπτωση/34.000 στο Ισραήλ, 1/78,000 έως 1/111.000 γεννήσεις αρρένων στη Βρεττανική Κολομβία (Lowry RB et al, 1990), 1/132. 000 στην Αγγλία (Schaap T and Bach G, 1980; Young ID and Harper PS, 1983; Lowry RB et al, 1990) και 1/72.000 γεννήσεις ζώντων αρρένων, στη Βόρεια Ιρλανδία (Nelson J, 1997).
Σύμφωνα με πρόσφατες μελέτες στη Γερμανία και την Ολλανδία, εκτιμάται σε 1 περίπτωση/140.000-330.000 γεννήσεις ζώντων παιδιών και 1.3 περιπτώσεις/100.000 γεννήσεις αρρένων (Wraith JE et al, 2008)
.
Φυλή. Το σύνδρομο Hunter απαντάται σε όλες τις φυλές, αν και φαίνεται ότι είναι συχνότερο σε Ιουδαίους που ζουν στο Ισραήλ (Schaap T and Bach G, 1980; Zlotogora J et al, 1991).
Φύλο. Αν και Χ-φυλοσύνδετο νόσημα, το σύνδρομο Hunter έχει αναφερθεί σποραδικά και στα θήλεα (Mossman J et al, 1983; Young ID and Harper PS, 1983; Broadhead DM et al, 1986; Clarke JT et al, 1991; Winchester B et al, 1992; Sukegawa K et al, 1997; Demitsu T et al, 1999).
Ηλικία. Ο βαρύς τύπος συνδρόμου Hunter διαγιγνώσκεται συνήθως στο 2ο-4ο έτος της ηλικίας. Ο ηπιότερος τύπος μπορεί να παραμείνει αδιάγνωστος μέχρι την εφηβική ηλικία ή ακόμα και στην ενήλικη ζωή.
ΑΙΤΙΟΛΟΓΙΑ
Το σύνδρομο Hunter οφείλεται σε μεταλλάξεις του γονιδίου το οποίο κωδικοποιεί την ιδουρονάτη-2-σουλφατάση (IDS) και εντοπίζεται στο χρωμόσωμα Xq28 (Sukegawa K et al, 1998). Η ανεπάρκεια της IDS οδηγεί σε εναπόθεση βλεννοπολυσακχαριδών στους ιστούς και απέκκριση μεγάλων ποσοτήτων θειικής ηπαριτίνης και θειικής χονδροϊτίνης Β από τα ούρα. Στο 50% περίπου των ινοβλαστών ανευρίσκονται μεταχρωματικά κυτταροπλασματικά έγκλειστα (Danes BS and Bearn AG, 1965).
ΚΛΙΝΙΚΕΣ ΕΚΔΗΛΩΣΕΙΣ
Κλινικά, το σύνδρομο Hunter διακρίνεται σε 2 τύπους :
1. Βαρύς τύπος (τύπος Α)
2. Ηπιότερος τύπος (τύπος Β)
Οι τύποι αυτοί αντιπροσωπεύουν τα 2 άκρα του εύρους της κλινικής βαρύτητας του συνδρόμου Hunter.
ΔΙΑΦΟΡΕΣ MPS ΤΥΠΟΥ Α ΑΠΟ MPS ΤΥΠΟΥ Β
1. Τύπος Β (ηπιότερος)
Ο τύπος αυτός παρουσιάζεται αργότερα στην εφηβική ή στην ενήλικη ζωή. Οι ασθενείς με MPS τύπου Β έχουν παρόμοιες κλινικές εκδηλώσεις με την MPS τύπου Α (τράχυνση των χαρακτηριστικών του προσώπου, βαρηκοΐα, δυσκαμψία των αρθρώσεων, προσβολή ανώτερων αναπνευστικών οδών, σύνδρομο καρπιαίου σωλήνα), οι οποίες όμως εξελίσσονται με βραδύτερο ρυθμό, αλλά δεν πάσχουν από διανοητική καθυστέρηση.
Οι ενήλικες με MPS II τύπου Β μπορεί ακόμα να έχουν σμίκρυνση των οστών του καρπού ή ήπια δυσπλασία της λεκάνης και των μηριαίων κεφαλών με πρόωρη οστεοαρθρίτιδα.
Στην MPS II τύπου Β, το ΚΝΣ προσβάλλεται ελάχιστα ή καθόλου, αν και οι επιπτώσεις της άθροισης των γλυκοζαμινογλυκανών στα άλλα οργανικά συστήματα μπορεί να είναι εξίσου σοβαρή με των ασθενών που έχουν προοδευτική εξασθένηση της αναγνώρισης.
Επιβίωση στα πρώιμα ενήλικα χρόνια με φυσιολογική νοημοσύνη είναι συχνή στην ομάδα αυτή.
2. Βαρύς τύπος (τύπος Α)
Ο βαρύς τύπος MPS II είναι 3-4 φορές συχνότερος από τον ήπιο. Χαρακτηρίζεται από ταχεία κινητική επιδείνωση και εξέλιξη των φυσικών παραμορφώσεων μετά το 3ο έτος της ζωής, με κακή κατάληξη μεταξύ 4ου-14ου έτους της ηλικίας. Το 84% των πασχόντων αρρένων μπορεί να έχει νευρολογικές ανωμαλίες και, στο 82%, προσβάλλεται το καρδιαγγειακό (Wraith JE et al, 2008).
Παρουσιάζεται συνήθως μεταξύ 2ου και 4ου έτους της ηλικίας με σκελετικές ανωμαλίες, βραχυσωμία, διανοητική καθυστέρηση, τράχυνση των χαρακτηριστικών του προσώπου και δυσκαμψία των αρθρώσεων (Young ID and Harper PS, 1983).
Αλλες εκδηλώσεις, με τις οποίες μπορεί να παρουσιασθεί η νόσος ή εμφανίζονται αργότερα, περιλαμβάνουν : υπερδραστηριότητα, προοδευτική απώλεια της ακοής, ηπατομεγαλία, σύνδρομο καρπιαίου σωλήνα, προοδευτική εκφύλιση του αμφιβληστροειδούς και υποτροπιάζουσες ωτικές λοιμώξεις.
Η προσβολή του ΚΝΣ, η σημαντικότερη εκδήλωση της MPS II τύπου Α, εκδηλώνεται κυρίως με προοδευτική ελάττωση της αναγνώρισης. Η ελάττωση της αναγνώρισης, σε συνδυασμό με προοδευτική πνευμονοπάθεια και καρδιοπάθεια, συνήθως οδηγεί στο θάνατο την 1η ή 2η δεκαετία της ζωής.
ΚΛΙΝΙΚΕΣ ΕΚΔΗΛΩΣΕΙΣ MPS II ΤΥΠΟΥ Α και Β
Αναπνευστικό.
Συχνές λοιμώξεις του ανώτερου αναπνευστικού είναι μία από τις πρωϊμότερες εκδηλώσεις της MPS II.
Οι αναπνευστικές οδοί προοδευτικά στενεύουν λόγω άθροισης GAG στη γλώσσα, τα μαλακά μόρια του στοματοφάρυγγα και την τραχεία, οδηγώντας σε αναπνευστική απόφραξη και άπνοια ύπνου, η οποία χρειάζεται θετική αναπνευστική υποστήριξη και τραχειοστομία.
Αρθρώσεις/σκελετός.
Συγκάμψεις των αρθρώσεων είναι μία από τις πρωϊμότερες εκδηλώσεις της νόσου. Αφορά ιδιαίτερα τις αρθρώσεις των φαλάγγων των χεριών και προκαλεί σημαντικό περιορισμό της κινητικότητας.
Οι σκελετικές ανωμαλίες χαρακτηρίζονται από πολλαπλή δυσόστωση, δηλ. γενικευμένη πάχυνση των μακρών οστών, ιδιαίτερα των πλευρών, και ακανόνιστη οστεοποίηση των επιφύσεων, και συχνά, εντομή των σπονδυλικών σωμάτων .
Η δυσπλασία των ισχίων είναι η συχνότερη σκελετική ανωμαλία και μπορεί να οδηγήσει σε σημαντική αναπηρία με πρόωρη οστεοαρθρίτιδα. Οι ενήλικες με MPS II τύπου Β παρουσιάζουν σμίκρυνση των οστων του καρπού ή ήπια δυσπλασία της λεκάνης και των μηριαίων κεφαλών με πρόωρη οστεοαρθρίτιδα. Η κινητικότητα των αρθρώσεων μειώνεται και τα δάκτυλα μπορεί να εμφανίσουν γαμψοδακτυλία.
Ο αυχένας είναι βραχύς. Ο θώρακας, πλατύς. Μπορεί να υπάρχει μέτρια θωρακοοσφυική κύφωση. Οι ασθενείς με MPS II έχουν δύσκαμπτο βάδισμα και, όταν βαδίζουν, λυγίζουν τον κορμό τους προς τα εμπρός. Ο κορμός είναι σχετικά βραχύτερος από τα μέλη.
Τα παιδιά με MPS II φαίνονται φυσιολογικά στη γέννηση. Τα πρώτα χρόνια της ζωής, το ύψος τους είναι συνήθως >50 εκατοστημόρια και σε μερικά, >97 εκατοστημόρια. Το ύψος των περισσότερων ασθενών με MPS II αρχίζει να ελαττώνεται μετά το 3ο έτος και, σε μερικούς, μετά το 5ο ή 6ο έτος της ηλικίας.
Κατά το 8ο έτος της ηλικίας, είναι <3ο εκατοστημόριο, και σχεδόν όλα τα παιδιά παρουσιάζουν αναστολή της ανάπτυξης πριν από την εφηβεία (Schulze-Frenking G et al, 2011). Το ύψος των ενηλίκων με ήπιο τύπο προσεγγίζει τα 120-140 cm (55), ενώ των ασθενών με βαρύ τύπο, τα 105-115 cm.
Γαστρεντερικό.
Οι περισσότεροι ασθενείς με MPS II παρουσιάζουν ηπατομεγαλία ή/ και σπληνομεγαλία. Ομφαλοκήλες/βουβωνοκήλες είναι επίσης συχνές. Στους νεότερους ασθενείς με σύνδρομο Hunter, η προσβολή του ΓΕΣ οδηγεί σε χρόνια διάρροια, λόγω δυσλειτουργίας του αυτόνομου νευρικού συστήματος και, πιθανώς, του γαστρεντερικού βλεννογόνου. Αργότερα, η διάρροια μπορεί να μετατραπεί σε σοβαρή δυσκοιλιότητα.
Δέρμα.
Βλατιδώδεις «μαργαριταροειδείς» δερματικές αλλοιώσεις παρατηρούνται και στους 2 τύπους της νόσου και έχουν τα εξής χαρακτηριστικά :
- Παρουσιάζονται τυπικά πριν από το 10ο έτος της ηλικίας και είναι ουσιαστικά παθογνωμονικές της νόσου (Scott HS et al, 1995)
- Εχουν χρώμα ελεφαντοστού και κατανέμονται συμετρικά στο άνω τμήμα της ράχης (μεταξύ της γωνίας της ωμοπλάτης και της πρόσθιας μασχαλιαίας γραμμής) και πάνω από την θωρακική περιοχή και το άνω–έξω τμήμα των βραχιόνων και των μηρών
- Ιστολογικά παρουσιάζουν δερματική βλεννίνωση
- Μογγολοειδείς κηλίδες (Mongolian spots) παρατηρούνται στην οσφυιοιερή περιοχή και είναι μεγάλες, επεκτεινόμενες προς τους γλουτούς και την ράχη. Υπερτρίχωση μπορεί να οδηγήσει σε συνοφρυδία.
Καρδιαγγειακό.
Καρδιαγγειακές ανωμαλίες είναι συχνές στα περισσότερα παιδιά με MPS II και η κύρια αιτία νοσηρότητας και θνητότητας. Το 82% των ασθενών αναπτύσσει καρδιαγγειακά σημεία/ συμπτώματα και το 62%, φυσήματα συνδεόμενα με βλάβες των καρδιακών βαλβίδων (μιτροειδούς, αορτής, τριγλώχινος και πνευμονικής) (ΠΙΝΑΚΑΣ 4).
ΠΙΝΑΚΑΣ 4. ΚΑΡΔΙΑΓΓΕΙΑΚΕΣ ΕΠΙΠΛΟΚΕΣ MPSII (Wraith et al, 2008)
ΚΑΡΔΙΑΓΓΕΙΑΚΗ ΠΡΟΣΒΟΛΗ ΕΠΙΠΟΛΑΣΜΟΣ
Προσβολή καρδιακών βαλβίδων 57%
Μυοκαρδιοπάθεια 8%
Ταχυκαρδία 7%
Υπέρταση 6%
Αρρυθμία και συμφορητική καρδιακή ανεπάρκεια 4%
Περιφερική αγγειοπάθεια 2%
Καρδιαγγειακό (βαρύς τύπος)
Η άθροιση των GAG στα φύλλα των καρδιακών βαλβίδων οδηγεί σε δυσλειτουργία της καρδιάς. Η προσβολή του μυοκαρδίου προκαλεί πάχυνση και συνακόλουθα οδηγεί σε στεφανιαία αρτηριοπάθεια, μυοκαρδιοπάθεια και, σε συνδυασμό με την αποφρακτική πνευμονοπάθεια, πνευμονική υπέρταση.
Νευρικό σύστημα.
Σε ασθενείς με ήπια MPS II, το ΚΝΣ προσβάλλεται ελάχιστα ή καθόλου, αν και οι επιπτώσεις της άθροισης GAG σε άλλα οργανικά συστήματα ενδέχεται να είναι εξίσου σοβαρή με τους ασθενείς που έχουν προοδευτική εξασθένηση της αναγνώρισης.
Η προσβολή του ΚΝΣ είναι η σημαντικότερη εκδήλωση των παιδιών με σοβαρή MPS II και εκδηλώνεται κυρίως με προοδευτική εξασθένηση της αναγνώρισης. Η εξασθένηση της αναγνώρισης, σε συνδυασμό με την προοδευτική αποφρακτική πνευμονοπάθεια και την καρδιακή προσβολή, συνήθως οδηγεί στο θάνατο κατά την 1η ή 2η δεκαετία της ζωής.
Οι συχνότερες νευρολογικές ανωμαλίες της MPS II είναι οι διαταραχές της συμπεριφοράς και της αναγνώρισης, στο 36% και 37% των ασθενών, αντίστοιχα (Wraith JE et al, 2008). Οι διαταραχές της συμπεριφοράς παρατηρούνται τόσο στον βαρύ, όσο και στον ήπιο τύπο της νόσου (Young ID and Harper PS, 1981; Wraith JE et al, 2008), αν και είναι συχνότερες στον βαρύ τύπο.
Καθυστέρηση των σφαιρικών αναπτυξιακών ορόσημων είναι τυπικά η πρώτη εκδήλωση της προσβολής του εγκεφάλου σε παιδιά με MPS II. Η εξέλιξη των εκδηλώσεων της προσβολής του ΚΝΣ είναι αμείλικτη, οδηγώντας συνήθως σε ύφεση της ανάπτυξης μεταξύ 6ου και 8ου έτους της ηλικίας.Οι άρρενες χωρίς προοδευτική προσβολή του ΚΝΣ έχουν φυσιολογική ή σχετικά φυσιολογική νοημοσύνη. Σπασμοί μπορεί επίσης να παρουσιασθούν. Στένωση του σπονδυλικού σωλήνα, ιδιαίτερα στην ΑΜΣΣ, με συμπίεση του νωτιαίου μυελού παρατηρείται συχνά.
Το σύνδρομο καρπιαίου σωλήνα (CTS) είναι συχνή επιπλοκή της MPS II. Σε αντίθεση με τους ενήλικες με CTS, τα περισσότερα παιδιά με MPS II δεν παρουσιάζουν τυπικά συμπτώματα παγιδευτικής νευροπάθειας. Πάντως, οι μελέτες της αγωγιμότητας των νεύρων είναι παθολογικές.
Επικοινωνών υδροκέφαλος δεν παρατηρείται συχνά σε ασθενείς με MPS II, αν και οίδημα της οπτικής θηλής έχει αναφερθεί χωρίς να υπάρχει αύξηση της ενδοκρανιακής πίεσης.
Οφθαλμοί. Οι ασθενείς με MPS II μπορεί να εμφανίσουν αμφιβληστροειδοπάθεια και απώλεια των οπτικών πεδίων. Το ηλεκτροαμφιβληστροειδογράφημα (ERG) μπορεί να δείξει δυσλειτουργία του αφιβληστροειδούς (Leung LS et al, 1971), αν και τα σημεία και συμπτώματα δεν σχετίζονται απαραίτητα με τις ERG αλλοιώσεις (Ashworth JL et al, 2006).
Σε αντίθεση με την MPS I, θολερότητες του κερατοειδούς παρατηρούνται σπάνια στη MPS II, αν και η εξέταση με σχισμοειδή λυχνία μπορεί να αποκαλύψει αλλοιώσεις του κερατοειδούς, οι οποίες όμως δεν επηρεάζουν την όραση.
Οίδημα της κεφαλής του οπτικού νεύρου παρατηρείται στο 20% και ατροφία του οπτικού νεύρου, στο 11% περίπου των ασθενών με MPS II (Collins ML et al, 1990; Ashworth JL et al, 2006).
Άλλες οφθαλμικές ανωμαλίες :
- Ατυπη εκφύλιση του αμφιβληστροειδούς
- Χρόνιο οίδημα της οπτικής θηλής, το οποίο ελαττώνει την όραση (Beck M, 1983; Beck M and Cole G, 1984; Caruso RC et al, 1986)
- Αμφοτερόπλευρες επαμφιβληστροειδικές μεμβράνες
Προσωπείο.
Το προσωπείο των ασθενών με τον βαρύ τύπο MPS II είναι συνήθως περισσότερο εντυπωσιακό από των ασθενών με ήπιο τύπο. Αρχικά, τα παιδιά με MPS II γεννιούνται φυσιολογικά στην όψη. Τράχυνση των χαρακτηριστικών του προσώπου γενικά παρουσιάζεται μεταξύ 18ου μηνός και 4ου έτους της ηλικίας στον βαρύ τύπο και περίπου 2 χρόνια αργότερα, στον ήπιο τύπο. Μακροκεφαλία έχουν όλοι οι ασθενείς με MPS II.
Οι ανωμαλίες των χαρακτηριστικών του προσώπου είναι αποτέλεσμα μακρογλωσσίας, αποπλάτυνσης της ρινός, προβολής των υπερόφρυων ακρολοφιών και εναπόθεσης GAGs στα μαλακά μόρια του προσώπου, η οποία οδηγεί σε μεγάλες κυκλοτερείς παρειές και παχειά χείλη.
Στόμα.
Οι συχνότερες στοματικές ανωμαλίες σε παιδιά με MPS II περιλαμβάνουν μακρογλωσσία και διόγκωση της γλώσσας (εντονότερη σε παιδιά ηλικίας >5 ετών), υπερτροφία των αδενοειδών εκβλαστήσεων και αγκύλωση της κροταφογναθικής άρθρωσης, η οποία περιορίζει το άνοιγμα του στόματος. Οι ανωμαλίες αυτές προκαλούν προοδευτικά δυσκολία στην κατάποση. Η εναπόθεση GAG στον λάρυγγα τυπικά δημιουργεί χαρακτηριστική τραχειά φωνή.
Οδόντες. Οι οδόντες συχνά έχουν ακανόνιστο σχήμα και φυτρώνουν αραιά και τα ούλα είναι υπερτροφικά. Ενίοτε παρατηρούνται οδοντοφόρες κύστεις, οι οποίες συχνά προκαλούν πόνο και ενόχληση και δύσκολα διαγιγνώσκονται, ιδιαίτερα σε άρρενες με προσβολή του ΚΝΣ.
Ωτα.
Οι περισσότεροι ασθενείς τόσο με MPS II τύπου Α όσο και Β, παρουσιάζουν προοδευτική απώλεια της ακοής, η οποία συνήθως είναι μικτού τύπου, αλλά μπορεί να είναι και νευροαισθητήρια ή αγώγιμη (Al Sawaf S et al, 2008). Ωτοσκλήρυνση μπορεί να συμβάλει στην αγώγιμη απώλεια της ακοής. Η νευροαισθητήρια απώλεια της ακοής αποδίδεται σε συμπίεση του κοχλιακού νεύρου οφειλόμενη σε αραχνοειδή υπερπλασία, ελάττωση του αριθμού των γαγγλιακών κυττάρων και εκφύλιση των τριχωτών κυττάρων.
ΚΛΙΝΙΚΕΣ ΕΚΔΗΛΩΣΕΙΣ MPS II ΚΑΙ ΤΩΝ 2 ΤΥΠΩΝ (Α και Β) (ΣΥΝΟΠΤΙΚΑ)
Αισθητήρια όργανα
- Απώλεια της ακοής (νευροαισθητήρια ή αγωγιμότητας, αλλά συνήθως μικτού τύπου)
- Προοδευτική εκφύλιση του αμφιβληστροειδούς και χρόνιο οίδημα της οπτικής θηλής, το οποίο οδηγεί σε απώλεια της όρασης
- Υποτροπιάζουσα μέση ωτίτιδα
Αναπνευστικό σύστημα
- Αποφρακτική πνευμονοπάθεια και αναπνευστική ανεπάρκεια
- Υποτροπιάζουσα ρινόρροια
Δέρμα- εξαρτήματα
- Βλατιδώδεις «μαργαριταροειδείς» δερματικές αλλοιώσεις χρώματος ελεφαντοστού
- Βλατιδώδες εξάνθημα στην περιοχή των ωμοπλατών και του δελτοειδούς
- Μογγολοειδείς κηλίδες στην περιοχή των γλουτών, της οσφύος και του ιερού, την ράχη και τον κορμό
- Πάχυνση του δέρματος
- Υπερτρίχωση
Θώρακας
- Πλατύς/κωνοειδής θώρακας
Κεφαλή και αυχένας
- Ανοιχτό στόμα
- Αραιή οδοντοφυία
- Βραχυλαιμία
- Διόγκωση της γλώσσας
- Μακροκεφαλία
- Πάχυνση των μυκτήρων και των χειλέων
- Σκαφοειδοκεφαλία
- Τράχυνση των χαρακτηριστικών του προσώπου
- Χαμηλή ρινική γέφυρα
Κοιλιά
- Ομφαλοκήλες ή βουβωνοκήλες
- Προβολή κοιλιάς
Μυοσκελετικό
- Βραχυσωμία
- Γαμψοδακτυλία
- Δύσκαμπτο βάδισμα
- Θωρακο-οσφυική κύφωση
- Κοιλοποδία
- Περιορισμός κινητικότητας των αρθρώσεων
- Πολλαπλή δυσόστωση
- Συγκάμψεις αγκώνων και ώμων
- Σύνδρομο καρπιαίου σωλήνα
- Υπερτονία
- Υπερδραστήρια μυϊκά αντανακλαστικά
Νευρικό σύστημα
- Αύξηση της ενδοκρανιακής πίεσης, η οποία οδηγεί σε ασυνήθιστη διόγκωση της κεφαλής (επικοινωνών υδροκέφαλος)
- Σοβαρή διανοητική καθυστέρηση
- Σπαστική τετραπληγία, λόγω πρόσκρουσης των παχυσμένων μηνίγγων στο νωτιαίο μυελό της ΑΜΣΣ (Ballenger CE et al, 1980)
- Σπασμοί
- Υπερδραστηριότητα και εκρηκτική, καταστρεπτική συμπεριφορά, παρόμοια με την παρατηρούμενη στο σύνδρομο San filippo (Zhao HG et al, 1996).
Ηπατομεγαλία-σπληνομεγαλία.
ΕΡΓΑΣΤΗΡΙΑΚΑ ΕΥΡΗΜΑΤΑ
Οι περισσότεροι ασθενείς με MPS II απεκκρίνουν περίπου ίσα ποσά DS και HS από τα ούρα
ΙΣΤΟΛΟΓΙΚΑ ΕΥΡΗΜΑΤΑ
Τα περιφερικά λεμφοκύτταρα δείχνουν μεταχρωματικά κοκκία με κενοτόπια. Τα περιφερικά κοκκιοκύτταρα και τα κύτταρα του μυελού των οστων μπορεί να δείξουν κοκκία Alder-Reilly. Στη βιοψία του επιπεφυκότα, το ηλεκτρονικό μικροσκόπιο δείχνει σχηματισμούς συνδεδεμένους με την μεμβράνη.
ΔΙΑΓΝΩΣΗ
Η διάγνωση και των 2 τύπων MPS II στους ημιζυγώτες μπορεί να γίνει με την ανεύρεση της ανεπάρκειας της IDU στον ορό, σε μεμονωμένα λευκά αιμοσφαίρια και σε καλλιεργημένους ινοβλάστες .
ΔΙΑΦΟΡΙΚΗ ΔΙΑΓΝΩΣΗ
- Βλεννοπολυσακχαρίδωση τύπου I H/S
- Βλεννοπολυσακχαρίδωση τύπου IH
- Βλεννοπολυσακχαρίδωση τύπου III
- Βλεννοπολυσακχαρίδωση τύπου IS
- Βλεννοπολυσακχαρίδωση τύπου VII
- Πολλαπλή ανεπάρκεια σουλφατάσης
ΘΕΡΑΠΕΙΑ
1. Ενζυμική θεραπεία αναπλήρωσης (ERT)
Idursulfase (elaprase) :
- Είναι ένας καθαρμένος τύπος ανθρώπινης ιδουρονικής σουλφατάσης
- Υδρολύει τους 2-θειικούς εστέρες των τελικών υπολειμματικών IDS από την θειική δερματάνη και θειική ηπαράνη στα λυσοσώματα των διαφόρων κυτταρικών τύπων
- Αναπληρώνει την ανεπάρκεια της ιδουρονάτης-2-σουλφατάσης
Ενδείξεις : MPS II (σύνδρομο Hunter)
Αποτελεσματικότητα :
- Βελτιώνει τον χρόνο βάδισης και μειώνει τον όγκο του ήπατος και του σπληνός και τα επίπεδα των γλυκοζαμινογλυκανών στα ούρα (Mullen CA et al, 2000; Martin R et al, 2008)
- Δεν εισδύει στο ΚΝΣ και δεν επηρεάζει την λειτουργία της αναγνώρισης, γι΄ αυτό και ο ρόλος της στη θεραπεία της MPS II είναι υπό αμφισβήτηση (Muenzer J et al, 2007)
Δόσεις :
- Ενήλικες :
- 0.5 mg/kg IV qwk σε διάστημα 1-3 ωρών
- Εάν υπάρξουν αντιδράσεις στην έγχυση, η έγχυση μπορεί να διαρκέσει έως και 8 ώρες
- Παιδιά :
- <5 ετών : Δεν έχει προσδιορισθεί
2. Μεταμόσχευση μυελού των οστών (BMT)
- Μπορεί να ελαττώσει την τράχυνση των χαρακτηριστικών του προσώπου και των τριχών της κεφαλής και να αυξήσει το εύρος της κίνησης των αρθρώσεων
- Δεν φαίνεται να βελτιώνει ή καθυστερεί την διανοητική καθυστέρηση και τις ανωμαλίες της ακοής. Σε μία μελέτη, από τα 16 παιδιά με σύνδρομο Hunter που υποβλήθηκαν σε ΒΜΤ, τα 15 παρουσίασαν σημαντική επιδείνωση της διανοητικής καθυστέρησης (Vellodi A et al, 1999)
3. Χειρουργική θεραπεία
- Χειρουργικές επεμβάσεις για τον χρόνιο υδροκέφαλο, το σύνδρομο καρπιαίου σωλήνα, τις κοιλιοκήλες, τις συγκάμψεις των αρθρώσεων
- Τραχειοστομία
ΠΡΟΓΝΩΣΗ
Οι ασθενείς με τον βαρύ τύπο συνήθως πεθαίνουν κατά το 10ο-15ο έτος της ηλικίας λόγω καρδιοαναπνευστικής ανεπάρκειας δευτεροπαθώς σε απόφραξη των ανώτερων αναπνευστικών οδών και κυρίως διηθητικής βαλβιδοπάθειας. Αντιθέτως, οι ασθενείς με ηπιότερους τύπους επιβιώνουν μέχρι την 6η και 7η δεκαετία της ζωής.
3. ΒΛΕΝΝΟΠΟΛΥΣΑΚΧΑΡΙΔΩΣΗ ΤΥΠΟΥ ΙΙΙ (Σύνδρομο Sanfilippo) (Mucopolysaccharidosis III; MPS III; Sanfilippo syndrome)
ΧΑΡΑΚΤΗΡΙΣΤΙΚΑ ΝΟΣΟΥ
Η MPS τύπου III (ή σύνδρομο Sanfilippo) χαρακτηρίζεται από βαριά διανοητική και νευρολογική εκφύλιση συνδεόμενη με σχετικά ήπιες εκδηλώσεις MPS συνεπεία προοδευτικής άθροισης περίσσειας θειικής ηπαράνης στα λυσοσώματα διαφόρων ιστών και οργάνων (Vitry S et al, 2009). Ταυτόχρονα, η θειική ηπαράνη απεκκρίνεται σε μεγάλα ποσά από τα ούρα.
ΕΠΙΔΗΜΙΟΛΟΓΙΑ
Συχνότητα. Στη Βόρεια Ιρλανδία η συχνότητα του συνδρόμου Sanfilippo υπολογίζεται σε 1 περίπτωση/280.000 γεννήσεις ζώντων παιδιών (0.36 περιπτώσεις/100.000 γεννήσεις ζώντων παιδιών) (Nelson J, 1997).
Η MPS IIIA είναι συχνότερη στις ΗΠΑ και την Αγγλία, ενώ η MPS IIIB, στην Ελλάδα (Beratis NG et al, 1986; Toone JR and Applegarth DA, 1988).
Στην Ολλανδία, μία μελέτη υπολόγισε την συχνότητα όλων συνολικά των πολυσακκχαριδώσεων σε 4.5 περιπτώσεις/100. 000 γεννήσεις ζώντων παιδιών (Poorthuis BJ et al, 1999). Σύμφωνα με την μελέτη αυτή, η MPS III αποτελεί το 47% όλων των διεγνωσμένων μέχρι σήμερα περιπτώσεων βλεννοπολυσακκχαρίδωσης και η επίπτωσή της είναι 1.89 περιπτώσεις/ 100,000 γεννήσεις ζώντων παιδιών. Στη μελέτη αυτή, η επίπτωση της βλεννοπολυσακκχαρίδωσης IIIA ήταν 1.16 περιπτώσεις/100,000 γεννήσεις ζώντων παιδιών.
Κατά τον Meikle και συν. (1999), η επίπτωση της βλεννοπολυσακχαρίδωσης τύπου ΙΙΙΑ ανέρχεται σε 1 περίπτωση/114,000 γεννήσεις ζώντων παιδιών και της βλεννοπολυσακχαρίδωσης τύπου ΙΙΙΒ, σε 1 περίπτωση/211,000 γεννήσεις ζώντων παιδιών (Meikle PJ et al, 2009).
Η συχνότητα της βλεννοπολυσακκχαρίδωσης τύπου IIIC και IIID είναι πολύ μικρότερη (1/1.407.000 και 1/1.056.000 γεννήσεις ζώντων παιδιών αντίστοιχα). Στην Αυστραλία (2000), η συχνότητα όλων των τύπων του συνδρόμου Sanfilippo έχει υπολογισθεί σε 1 περίπτωση/66.000 γεννήσεις ζώντων παιδιών.
Φυλή. Οι βλεννοπολυσακκχαριδώσεις δεν έχουν φυλετική προτίμηση.
Φύλο. Οι MPS τύπου ΙΙΙ μεταβιβάζονται σύμφωνα με τον αυτοσωμικό υπολειπόμενο Μενδέλειο τύπο. Τα μεταλλαγμένα γονίδια εντοπίζονται στα αυτοσώματα, και όχι στα φυλετικά χρωμοσώματα, γι΄αυτό και το σύνδρομο Sanfilippo προσβάλλει άρρενες σε ίση συχνότητα με τις θήλεις.
ΤΥΠΟΙ ΣΥΝΔΡΟΜΟΥ SANFILIPPO
Η MPS III διακρίνεται σε 4 τύπους, καθένας εκ των οποίων οφείλεται σε ανεπάρκεια διαφορετικού ενζύμου (Zhang WM et al, 2008) :
1. Τύπος IIIA (σύνδρομο Sanfilippo A) (type IIIA; Sanfilippo A) (McKusick 252900)
- Ανεπάρκεια heparan N-sulfatase (17q25.3)
2. Τύπος IIIB (σύνδρομο Sanfilippo B) (type IIIB; Sanfilippo B) (McKusick 252920)
- Ανεπάρκεια N-alpha-acetylglucosaminidase (17q21)
3. Τύπος IIIC (σύνδρομο Sanfilippo C) (type IIIC; Sanfilippo C) (McKusick 252930)
- Ανεπάρκεια heparin acetyl-CoA:alpha-glucosaminide N-acetyltransferase (8p11.1)
4. Τύπος IIID (σύνδρομο Sanfilippo D) (type IIID; Sanfilippo D) (Mc Kusick 252940)
- Ανεπάρκεια N-acetylglucosamine-6-sulfatase (12q14)
ΑΙΤΙΟΛΟΓΙΑ
Η MPS τύπου ΙΙΙ μεταδίδεται σύμφωνα με το αυτοσωμικό υπολειπόμενο πρότυπο κληρονομικότητας. Η ανεπάρκεια 4 διαφορετικών ενζύμων ευθύνεται για τους διάφορους τύπους του συνδρόμου Sanfilippo.
Οι τύπος ΙΙΙΑ οφείλεται σε έλλειψη της σουλφατάσης της θειικής ηπαράνης (heparan-N-sulfate) (McDowell GA et al, 1993; Scott HS et al, 1995)
Ο τύπος ΙΙΙΒ οφείλεται σε έλλειψη του ενζύμου Ν-ακετυλ-α-D-γλυκοζαμινιδάση) (Ν -acetyl-alpha-D-glucosaminidase; NAG) (Ohmi K et al, 2009)
Ο τύπος ΙΙΙC οφείλεται σε έλλειψη του ενζύμου ακετυλ-CoA:α-γλυκοζαμινική ακετυλτρανσφεράση (acetyl-CoA:alpha- glucosaminide acetyltransferase) (Klein U et al, 1981)
Ο τύπος ΙΙΙD οφείλεται σε έλλειψη του ενζύμου Ν- ακετυλγλυκοζαμινο-6-σουλφατάση (N-acetylglucosamine-6-sulfatase) (Coppa GV et al, 1983).
Οι γονιδιακές μεταλλάξεις των 4 αυτών τύπων του συνδρόμου Sanfilippo έχουν χαρτογραφηθεί στο ανθρώπινο γονιδίωμα και εντοπίζονται στα εξής χρωμοσώματα
- Τύπος IIIA : 17q25.3
- Τύπος IIIB : 17q21 (NAGLU) (Zhao HG et al, 1995; Zhao HG et al, 1996)
- Τύπος IIIC : 8p11.1
- Τύπος IIID : 12q14
ΚΛΙΝΙΚΗ ΕΙΚΟΝΑ
Οι πάσχοντες από MPS IIIA γενικά προσβάλλονται βαρύτερα από τους πάσχοντες από MPS IIIΒ ή MPS IIIC (Van de Kamp JJP et al, 1981; Van Schrojenstein-de Valk HMJ and van de Kamp JJPl, 1987; Lindor NM et al, 1994; Wraith JE, 1995; Aronovich EL et al, 2000)
.
Η MPS IIIA έχει πρωϊμότερη έναρξη, είναι περισσότερο έκδηλη και οδηγεί πρόωρα στο θάνατο.
Οι περισσότεροι ασθενείς με σύνδρομο Sanfilippo γεννιούνται χωρίς συμπτώματα και αναπτύσσονται κανονικά τα 2 πρώτα χρόνια της ζωής. Κατά το 2ο όμως έτος της ζωής, η ανάπτυξη ανακόπτεται και οι ασθενείς παρουσιάζουν διαταραχές της συμπεριφοράς, όπως ανησυχία, επιθετικότητα, μείωση του εύρους της προσοχής, δυσκολία στην άρθρωση του λόγου και διαταραχές του ύπνου.
Η επιθετική υπερδραστηριότητα είναι συχνά η αιτία για την οποία οι γονείς ζητούν ιατρική βοήθεια και εντονότερη στα θήλεα, παρά τα άρρενα. Λόγω αυτής της υπερδραστηριότητας, τα πάσχοντα παιδιά χάνουν προοδευτικά τις διανοητικές και κινητικές τους δεξιότητες.
Η απώλεια της επαφής με το περιβάλλον είναι εμφανής, πριν οι ασθενείς γίνουν «φυτά», με σπαστική διπληγία και καταλήξουν κακώς περί το 10ο-20ο έτος της ηλικίας (Nidiffer FD and Kelly TE, 1983).
Τα πρώτα σχολικά χρόνια τα συμπτώματα επιδεινώνονται, και οι πάσχοντες έχουν περιορισμένη ικανότητα παρακολούθησης και έλλειψη συγκέντρωσης. Συχνά ακόμα παρουσιάζουν υποτροπιάζουσες λοιμώξεις του ανώτερου αναπνευστικού και αναπνευστική ανεπάρκεια οφειλόμενη σε απόφραξη των αναπνευστικών οδών λόγω των ανατομικών ανωμαλιών, νευρολογικής εξασθένησης και αύξησης των εκκρίσεων
Σοβαρό πρόβλημα στα μικρά παιδιά συχνά είναι υποτροπιάζουσα ή χρόνια διάρροια, η οποία όμως υποχωρεί με την πάροδο της ηλικίας. Η διάρροια οφείλεται μάλλον στη λυσοσωμική εναπόθεση γλυκοζαμινογλυκανών στους νευρώνες του μυεντερικού πλέγματος.
Η νοητική ανάπτυξη επιβραδύνεται μετά την νηπιακή ηλικία. Υστερα, η βάδιση και η άρθρωση του λόγου προοδευτικά επιδεινώνονται. Μερικοί ασθενείς ουδέποτε μιλούν. Οι περισσότεροι ασθενείς με σύνδρομο Sanfilippo A παρουσιάζουν σοβαρή διανοητική καθυστέρηση κατά το 6ο έτος της ηλικίας.
Διαταραχές του ύπνου είναι πολύ συχνές. Ηπατοσπληνομεγαλία παρατηρείται σε >80% των ασθενών. Οι εντυπωσιακές κλινικές εκδηλώσεις (τράχυνση των χαρακτηριστικών του προσώπου, σκελετικές ανωμαλίες), οι οποίες παρατηρούνται στις άλλες βλεννοπολυσακκχαριδώσεις, δεν είναι τόσο έντονες στους ασθενείς με σύνδρομο Sanfilippo.
Το ύψος των ασθενών με MPS III είναι φυσιολογικό ή ελαφρώς χαμηλότερο του φυσιολογικού. Η κινητικότητα των αγκώνων και των γονάτων μπορεί να είναι περιορισμένη.
Ακτινολογικά, παρατηρείται πάχυνση του πρόσθιου τμήματος του θόλου του κρανίου, σκλήρυνση των μαστοειδών αποφύσεων, σπονδυλικά σώματα ωοειδούς σχήματος και ήπια υποπλασία των λαγονίων ύπερθεν των κοτυλών. Η πολλαπλή δυσόστωση αναπτύσσεται αργά και είναι πολύ ήπια.
Κατά το 10ο έτος της ηλικίας, συνήθως αναπτύσσουν σοβαρό περιορισμό των δραστηριοτήτων και της κινητικότητας.
Τα μικρά παιδιά δεν παρουσιάζουν ανωμαλίες ή δυσμορφίες του προσώπου. Τα μεγαλύτερα παιδιά μπορεί να εμφανίσουν ήπια τράχυνση των χαρακτηριστικών του προσώπου, παρόμοια με την παρατηρούμενη σε ασθενείς με φαινότυπο MPS I-H. Περίπου 80% των παιδιών με MPS III έχει όψη διανοητικά καθυστερημένου ατόμου, με ελαφρώς εμβυθισμένη ρινική γέφυρα, και πλούσια και τραχιά μαλλιά.
Οι τρίχες των μαλλιών μπορεί να είναι τριγωνικές (Crump IA and Danks DM, 1971), ή να έχουν μεγάλη ποικιλομορφία στο σχήμα και το χρώμα (Teschler-Nicola M, Killian W, 1982).
Θολερότητες κερατοειδούς απουσιάζουν.
Σπασμοί μπορεί να εμφανισθούν, αλλά συνήθως ελέγχονται εύκολα με την φαρμακευτική αγωγή.
Στα όψιμα στάδια της νόσου οδοντικά αποστήματα είναι σημαντικό πρόβλημα. Η γλώσσα δεν είναι μεγάλη, αλλά μπορεί να προβάλλει στα όψιμα στάδια της νόσου.
ΚΛΙΝΙΚΕΣ ΕΚΔΗΛΩΣΕΙΣ (ΣΥΝΟΠΤΙΚΑ)
Αισθητήρια όργανα
- Απώλεια ακοής
- Δυσλειτουργία οφθαλμικών βολβών
- Θολερότητες κερατοειδούς
- Καταράκτες
- Μυωπία
Αναπνευστικές
- Υποτροπιάζουσες ανώτερες αναπνευστικές λοιμώξεις
Γαστρεντερικές
- Ηπια ηπατομεγαλία και σπληνομεγαλία
- Σοβαρή διάρροια/δυσκοιλιότητα
Νευρολογικές
- Ανοια
- Απώλεια ακοής και καθυστέρηση ομιλίας (σε βαρέως πάσχοντες)
- Αταξία
- Ελάττωση της διάρκειας της προσοχής
- Επιθετική και καταστρεπτική συμπεριφορά
- Διανοητική καθυστέρηση
- Διαταραχές του ύπνου
- Δυσκολία στην άρθρωση του λόγου
- Σπασμοί
- Τρόμος
- Υπερδραστηριότητα
- Σοβαρή προοδευτική νευρολογική εκφύλιση
Πρόσωπο
- Ηπια τράχυνση των χαρακτηριστικών του προσώπου
- Συνοφρυδία
Σκελετικές ανωμαλίες
- Ανωμαλία μεγέθους/σχήματος σπονδύλων
- Ανωμαλίες πλευρών
- Ανωμαλία/απουσία κλείδας
- Βλαισογωνία
- Βραχυσωμία
- Δυσκαμψία και περιορισμός της κινητικότητας των αρθρώσεων
- Εξάρθρημα ισχίου
- Ηπια πολλαπλή δυσόστωση
- Μεγαλοκεφαλία
- Πάχυνση/πύκνωση κρανίου
- Ραιβοποδία/βλαισοποδία
- Σκολίωση
Αλλες
- Τραχιές/παχειές τρίχες μαλλιών
- Κήλες (βουβωνοκήλη ή/και ομφαλοκήλη)
ΔΙΑΓΝΩΣΗ
Η οριστική διάγνωση των υπότυπων της MPS III, η εντόπιση των φορέων και η προνεογνική διάγνωση μπορεί να γίνει με την ανεύρεση του ανεπαρκούντος ενζύμου στις λάχνες του χορίου ή σε καλλιεργημένα αμνιοκύτταρα (Matalon R et al, 1988; Applegarth DA, 1988; Nowakowski RW et al, 1989; Toone JR Di Natale P et al, 1991; Kleijer WJ et al, 1996).
ΔΙΑΦΟΡΙΚΗ ΔΙΑΓΝΩΣΗ
- Βλεννοπολυσακκχαρίδωση τύπου I H/S
- Βλεννοπολυσακκχαρίδωση τύπου IH
- Βλεννοπολυσακκχαρίδωση τύπου II
- Βλεννοπολυσακκχαρίδωση τύπου IS
- Βλεννοπολυσακκχαρίδωση τύπου IV
- Βλεννοπολυσακκχαρίδωση τύπου VI
- Βλεννοπολυσακκχαρίδωση τύπου VII
ΘΕΡΑΠΕΙΑ
Δεν υπάρχει οριστική θεραπεία του συνδρόμου Sanfilippo. Η θεραπεία είναι συμπτωματική και υποστηρικτική.
Μεταμόσχευση μυελού οστών (BMT). Η BMT βελτιώνει την κλινική έκβαση των ασθενών με βλεννοπολυσακκχαριδώσεις, κυρίως Ι, ΙΙ και VI, αν και δεν φαίνεται να έχει αποτέλεσμα στην βλεννοπολυσακκχαρίδωση III.
Ενζυμική θεραπεία αναπλήρωσης (ERT). Εχει εγκριθεί στις ΗΠΑ για την θεραπεία της βλεννοπολυσακκχαρίδωσης Ι, ΙΙ και VI. Στην βλεννοπολυσακκχαρίδωση IIIA και IIIB δεν φαίνεται να βελτιώνει την πρόγνωση, δεδομένου ότι τα ανασυνδυασμένα ένζυμα δεν μπορούν να διαπεράσουν τον αιματοεγκεφαλικό φραγμό και να μπούν στο ΚΝΣ.
Γονιδιακή θεραπεία. Η γονιδιακή θεραπεία της βλεννοπολυσακκχαρίδωσης τύπου IIIB μελετάται στις ΗΠΑ και την Γαλλία στα πειραματόζωα.
Φάρμακα :
- Αντισπασμωδικά (για τους σπασμούς)
- Ηρεμιστικά και μελατονίνη (για την βελτίωση της ποιότητας του ύπνου), σε συνδυασμό με αυστηρό πρόγραμμα νυχτερινής κατάκλισης σε ήσυχο, σκοτεινό δωμάτιο
ΠΡΟΓΝΩΣΗ
Το σύνδρομο Sanfilippo έχει προοδευτική πορεία με καταστροφική πρόγνωση. Με την πάροδο του χρόνου, οι ασθενείς αναπτύσσουν εκφύλιση του ΚΝΣ και μετατρέπονται σε «φυτά». Οι περισσότεροι ασθενείς δεν επιζούν πέραν του 20ού έτους της ηλικίας. Ο θάνατος οφείλεται κυρίως σε καρδιοαναπνευστική ανεπάρκεια οφειλόμενη σε αποφρακτική πνευμονοπάθεια ή/ και πνευμονικές λοιμώξεις.
Ο τύπος IIIA έχει την χειρότερη πρόγνωση. Οι περισσότεροι ασθενείς με τον τύπο αυτό πεθαίνουν στη διάρκεια της εφηβικής ηλικίας.
4. ΒΛΕΝΝΟΠΟΛΥΣΑΚΧΑΡΙΔΩΣΗ ΤΥΠΟΥ IV (Σύνδρομο Morquio) (Mucopolysaccharidosis IV; MPS IV; Morquio syndrome)
ΧΑΡΑΚΤΗΡΙΣΤΙΚΑ ΝΟΣΟΥ
To σύνδρομο Morquio (McKusick 253000) είναι γενετικό νόσημα του μεταβολισμού των βλεννοπολυσακχαριδών χαρακτηριζόμενο από :
- σκελετικές ανωμαλίες,
- αστάθεια των αρθρώσεων,
- αυχενική μυελοπάθεια και
- αυξημένη αποβολή θειικής κερατάνης (KS) από τα ούρα.
Οι ασθενείς με MPS IV συνήθως ξεχωρίζουν κλινικά από τους πάσχοντες από άλλες MPS, δοθέντος ότι δεν παρουσιάζουν τράχυνση των χαρακτηριστικών του προσώπου ή διανοητική καθυστέρηση και έχουν βαρύτερη προσβολή της ΣΣ, με σκολίωση, κύφωση, σοβαρή ύβωση, πλατυσπονδυλία και υποπλασία της οδοντοειδούς απόφυσης, όπως και αποπλάτυνση των πλευρών, τροπιδοειδή θώρακα, βραχυκορμικό νανισμό και βλαισογωνία και χαλάρωση των συνδέσμων. Η υποπλασία της οδοντοειδούς απόφυσης είναι η σημαντικότερη σκελετική ανωμαλία σε όλους τους ασθενείς με MPS IV.
ΤΥΠΟΙ ΣΥΝΔΡΟΜΟΥ MORQUIO
Το σύνδρομο Morquio διακρίνεται σε 2 (ή 3) τύπους :
1. Σύνδρομο Morquio, τύπος Α (MPS IVA) (βλεννοπολυσακχαρίδωση τύπου 4Α)
2. Σύνδρομο Morquio, τύπος Β (MPS IVB) (βλεννοπολυσακχαρίδωση τύπου 4Β)
3. Σύνδρομο Morquio, τύπος C (MPS IVC)
Υπάρχει και ένας 3ος τύπος συνδρόμου Morquio, ο τύπος C (252300), ο οποίος δεν χαρακτηρίζεται από αποβολή θειικής κερατάνης από τα ούρα.
ΑΙΤΙΟΛΟΓΙΑ
Το σύνδρομο Morquio τύπου Α (MPS IVA) οφείλεται σε ανεπάρκεια της γαλακτοζαμινο-6-σουλφατάσης (γονίδιο GALNS). Η ανεπάρκεια του ενζύμου αυτού οδηγεί σε ανεπαρκή αποδόμηση της θειικής κερατάνης, εναπόθεση
γλυκοζαμινογλυκανών (GAG) σε διάφορους ιστούς και αποβολή μεγάλων ποσοτήτων θειικής κερατάνης από τα ούρα.
Το σύνδρομο Morquio τύπου Β (MPS IVB) οφείλεται σε μεταλλάξεις του γονιδίου το οποίο κωδικοποιεί την β-γαλακτοσιδάση (GLB1; 611458) (Matalon R et al, 1974).
H MPS IVB είναι νόσημα αλληλικό με τους διάφορους τύπους γαγγλιοσίδωσης GM1, οι οποίοι χαρακτηρίζονται από προσβολή του ΚΝΣ.
Και οι δύο τύποι μεταβιβάζονται σύμφωνα με το αυτοσωμικό υπολειπόμενο πρότυπο κληρονομικότητας. Το υπεύθυνο γονίδιο εντοπίζεται στη χρωμοσωμική περιοχή 16q 24.3.
ΔΙΑΦΟΡΕΣ ΣΥΝΔΡΟΜΟΥ MORQUIO ΤΥΠΟΥ Α ΚΑΙ Β
Η MPS IVA διακρίνεται σε σοβαρούς, ενδιάμεσους και ήπιους τύπους, ένδειξη ότι στη γένεσή της εμπλέκονται διαφορετικά αλλήλια. Οι διαφορές στη βαρύτητα μπορεί επίσης να ερμηνευθούν από την ανεύρεση, σε μερικές περιπτώσεις, μειωμένης δραστηριότητας της νευραμινιδάσης (Glossl J et al, 1981). Σε αντίθεση με τους ασθενείς με MPS IVA, οι ασθενείς με MPS IVB έχουν σημαντικά ηπιότερο φαινότυπο :
- Ηπιότερη πολλαπλή δυσόστωση
- Ηπιο «θώρακα υποδηματοποιών»
- Θολερότητες του κερατοειδούς
- Υποπλασία της οδοντοειδούς απόφυσης
- Μέτρια οσφυική κύφωση και
- Ηπια βλαισογωνία.
Στην MPS IVB η υπερώα είναι συνήθως πλατειά και τα μεσοδόντια διαστήματα, αραιά. Σε αντίθεση με την MPS IVA, η αδαμαντίνη είναι φυσιολογική. Το εύρημα αυτό επιτρέπει την κλινική διάκριση μεταξύ των 2 αυτών τύπων.
Οι ασθενείς με MPS IVB αρχικά δεν παρουσιάζουν τράχυνση των χαρακτηριστικών του προσώπου, όπως στους άλλους τύπους MPS. Πάντως, η συνεχιζόμενη εναπόθεση θειικής κερατάνης στα κύτταρα μπορεί να οδηγήσει σε κάποια τράχυνση των χαρακτηριστικών του προσώπου και ηπατομεγαλία.
ΕΠΙΔΗΜΙΟΛΟΓΙΑ
Συχνότητα. Η συχνότητα του συνδρόμου Morquio υπολογίζεται σε 1 περίπτωση/ 75.000 γεννήσεις ζώντων παιδιών στη Βόρεια Ιρλανδία, 1/200.000, στη Βρεττανική Κολομβία και 1/263.157, στην Γερμανία.
Φυλή. Το σύνδρομο Morquio δεν έχει φυλετική προτίμηση.
Φύλο. Η αναλογία αρρένων/θήλεα είναι 1:1.
Ηλικία. Οι ασθενείς με σύνδρομο Morquio φαίνονται υγιείς στη γέννηση. Η διάγνωση της νόσου του γίνεται κατά το 6ο έτος της ηλικίας. Οι πάσχοντες συνήθως εξετάζονται για πρώτη φορά κατά το 2ο-3ο έτος της ηλικίας λόγω παραμορφώσεων της σπονδυλικής στήλης, αναστολής της ανάπτυξης και βλαισογωνίας.
Οι ασθενείς με ήπιες εκδηλώσεις συνδρόμου Morquio, ανεξαρτήτως τύπου, επιβιώνουν μέχρι την 7η δεκαετία της ζωής, σε αντίθεση με τους ασθενείς με σοβαρές εκδηλώσεις, κυρίως σχετιζόμενες με αυχενική αστάθεια, δεν ζουν τόσο πολύ.
ΚΛΙΝΙΚΗ ΕΙΚΟΝΑ ΣΥΝΔΡΟΜΟΥ MORQUIO
Οι κύριες κλινικές εκδηλώσεις του συνδρόμου Morquio προέρχονται από τον σκελετό και τις επιπτώσεις τους στο ΚΝΣ. Σε αντίθεση με τις περισσότερες βλεννοπολυσακκχαριδώσεις (MPS), το σύνδρομο Morquio δεν παρουσιάζεται με διανοητικές ανωμαλίες ή απώλεια των αναπτυξιακών ορόσημων, αλλά με ιστορικό αυξημένης αδεξιότητας και συχνών πτώσεων.
Οι κλινικές του εκδηλώσεις, όπως συμβαίνει σε όλες τις MPS, επιδεινώνονται με την πάροδο του χρόνου. Προοδευτικά, λόγω της συνεχιζόμενης εναπόθεσης θειικής κερατάνης στα λυσοσώματα ιστών και οργάνων, οι ασθενείς με MPS IV αναπτύσσουν ήπια τράχυνση των χαρακτηριστικών του προσώπου, θολερότητες του κερατοειδούς και ηπατοσπληνομεγαλία.
Υποπλασία οδοντοειδούς απόφυσης. Κύριο χαρακτηριστικό της MPS IV είναι η υποπλασία της οδοντοειδούς απόφυσης (Nelson J and Thomas PS, 1988). Η ανωμαλία αυτή, σε συνδυασμό με χαλάρωση των συνδέσμων, οδηγεί σε υπεξάρθρημα της ατλαντοαξονικής και αυχενική μυελοπάθεια, η οποία μπορεί να προκαλέσει διαταραχές της καρδιοαναπνευστικής λειτουργίας, τετραπάρεση ή ακόμα και θάνατο (Kopits SE, 1976; Tobias JD, 1999).
Αυχενική μυελοπάθεια μπορεί ακόμα να προκύψει από υπερτροφικά μαλακά μόρια αναπτυσσόμενα έμπροσθεν του νωτιαίου μυελού ισοϋψώς με το ανώτερο τμήμα της ΣΣ, λόγω εναπόθεσης πολυσακχαριδών (Herman MJ and Pizzutillo PD, 1999).
Οι ασθενείς με MPS IV και δυσανεξία στις ασκήσεις ενδέχεται να έχουν ασυμπτωματική αυχενική μυελοπάθεια, η οποία μπορεί να προκαλέσει δυσλειτουργία του εντέρου και της ουροδόχου κύστης.
Σκελετικές ανωμαλίες. Όπως συμβαίνει στις περισσότερες βλεννοπολυσακχαριδώσεις, οι πάσχοντες από σύνδρομο Morquio γεννιούνται με φυσιολογική σωματική διάπλαση, αλλά, στη διάρκεια των 2-3 πρώτων ετών της ζωής, παρουσιάζουν ασυνήθιστες μυοσκελετικές ανωμαλίες, όπως:
- Αναστολή της ανάπτυξης με βράχυνση του κορμού, της ΣΣ και του αυχένα
- Αρθρικές ανωμαλίες (υπερευλυγισία έως συγκάμψεις)
- Βλαισογωνία/ραιβογωνία
- Διαπλάτυνση των κατώτερων πλευρών
- Κύφωση
- Μυϊκή υποτονία και αδυναμία
- Σκολίωση
- Τροπιδοειδή θώρακα
- Υβωση της ΣΣ
Άλλες, λιγότερο συχνές, εκδηλώσεις :
- Ηπατομεγαλία
- Διαταραχές της ακοής
- Κήλες
- Παλινδρόμηση της αορτής ή/και της μιτροειδούς και υποπλασία της αδαμαντίνης
- με προδιάθεση για τερηδόνα.
ΚΛΙΝΙΚΕΣ ΕΚΔΗΛΩΣΕΙΣ ΣΥΝΔΡΟΜΟΥ MORQUIO (ΣΥΝΟΠΤΙΚΑ)
Κεφαλή και αυχένας
- Βραχυλαιμία
- Ηπια σκαφοκεφαλία-μακροκεφαλία
- Προγναθισμός
- Τράχυνση των χαρακτηριστικών του προσώπου (ηπιότερη του συνδρόμου Hunter ή Hurler)
Στόμα -οδόντες
- Ανωμαλίες των οδόντων με επιπέδωση και κοίλη διαμόρφωση των κονδύλων της γνάθου (τύπος IVA)
- Αποπλάτυνση υπερώας και διεύρυνση των μεσοδόντιων διαστημάτων (τύπος IVΒ)
- Λέπτυνση της αδαμαντίνης των οδόντων με φυσιολογική ακτινοπυκνότητα παρόμοια με την παρατηρούμενη στην υποπλαστική αμελογένεση (Barker D and Welbury RR, 2000)
- Μεγάλο στόμα
- Υποπλασία των οδόντων (Levin LS et al, 1975)
Θώρακας
- Αύξηση της προσθιοπίσθιας και ελάττωση της κάθετης διαμέτ-ρου του θώρακα
- Βράχυνση, πάχυνση και αποπλάτυνση των πλευρών με βολβώδη άκρα
- Διόγκωση της καρδιάς, λόγω προσβολής της αορτικής βαλβίδας
- Πρόωρη συνοστέωση και πρόσθια προβολή του στέρνου
- Τροπιδοειδής θώρακας
Αισθητήρια όργανα
- Γλαύκωμα (Cahane M et al, 1990; Gosele S et al, 2000)
- Διάχυτες θολερότητες κερατοειδούς/σκληροκερατοειδούς
- Νευροαισθητήρια κώφωση
Κοιλιά
- Βουβωνοκήλες
- Ηπατομεγαλία
- Προβολή κοιλιάς
Σπονδυλική στήλη
- Βράχυνση ΣΣ
- Κύφωση
- Προβολή ΟΜΣΣ
- Σκολίωση
- Υπερλόρδωση
Οστά-αρθρώσεις
- Βλαισότητα αγκώνων-γονάτων
- Επιμήκυνση των μελών
- Διόγκωση – ωλένια απόκλιση των καρπών
- Δυσκαμψία αρθρώσεων/αρθρίτιδα
- Κλίση του κάτω πέρατος της κερκίδας προς την ωλένη
- Πάχυνση των γονάτων
- Πλατυποδία
- Υπερευλυγισία των αρθρώσεων, δευτεροπαθώς σε χαλάρωση των συνδέσμων, ή περιορισμός της κινητικότητας μεγάλων αρθρώσεων (κυρίως ισχίων και γονάτων και ενίοτε αγκώνων)
Καρδιαγγειακό σύστημα
- Μυοκαρδιοπάθεια
- Παλινδρόμηση-ανεπάρκεια αορτής
ΑΚΤΙΝΟΛΟΓΙΚΑ ΕΥΡΗΜΑΤΑ
Λεκάνη – ισχία
- Αύξηση λοξότητας της έξω επιφάνειας της οροφής της κοτύλης
- Αποπλάτυνση και επιπέδωση των λαγόνιων οστών
- Διαπλάτυνση του αυχένα του μηριαίου με coxa valga
- Διεύρυνση της κοτύλης με προοδευτικό υπεξάρθρημα της μηριαίας κεφαλής
- Δυσπλασία της λεκάνης
- Επιπέδωση μηριαίων κεφαλών
Σπονδυλική στήλη
- Κυκλοτερής διαμόρφωση των σπονδυλικών σωμάτων, με κεντρική προεξοχή στην πρόσθια επιφάνειά τους. Παρατηρείται στη πρώιμη βρεφική ηλικία και είναι χαρακτηριστικό ακτινολογικό εύρημα της νόσου. Στους ενήλικες οι σπόνδυλοι είναι επίπεδοι και ορθογώνιοι με ανώμαλα όρια.
- Γλωσσοειδείς προσεκβολές ΟΜΣΣ (τύπος IVB)
- Θωρακοοσφυική κύφωση (τύπος IVB)
- Πλατυσπονδυλία (στο 2ο έτος της ηλικίας) και σπονδυλοεπιφυσιακή δυσπλασία
- Υπεξάρθρημα Α2-Α3 (τύπος IVB)
- Υποπλασία οδοντοειδούς απόφυσης (Arbisser et al, 1977)
Ανω – κάτω άκρα
- Βλαισογωνία (ήπια)
- Βράχυνση και αποπλάτυνση των μετακαρπίων και βράχυνση των φαλάγγων
- Ηπια διόγκωση των μετακαρπίων με όξυνση των περιφερικών άκρων
- Λοξή διαμόρφωση των περιφερικών αρθρικών επιφανειών της ωλένης και της κερκίδας
- Μικρά και ακανόνιστα οστά καρπού
Αλλα
- Βράχυνση των μακρών σωληνωδών οστών με διαπλάτυνση των μεταφύσεων και παραμόρφωση των επιφύσεων
- Επίπεδοι ή αμφίκοιλοι κόνδυλοι κάτω γνάθου (Rolling I et al, 1999)
- Οστεοπόρωση
- Πολλαπλή δυσόστωση (Arbisser AI et al, 1977)
- «Θώρακας υποδηματοποιών» (τύπος IVB)
- Πρόωρη σύγκλειση των ραφών του κρανίου
Τα ακτινολογικά αυτά ευρήματα, σε συνδυασμό με τις βιοχημικές διαταραχές, βοηθούν στη διάκριση του συνδρόμου Morquio από την σπονδυλοεπιφυσιακή δυσπλασία, η οποία έχει παρόμοια ακτινολογικά ευρήματα από την ΣΣ, αλλά συνδέεται συχνότερα με ραιβό ισχίο.
ΔΙΑΦΟΡΙΚΗ ΔΙΑΓΝΩΣΗ
- Βλεννοπολυσακχαρίδωση τύπου I H/S
- Βλεννοπολυσακχαρίδωση τύπου IH
- Βλεννοπολυσακχαρίδωση τύπου II
- Βλεννοπολυσακχαρίδωση τύπου III
- Βλεννοπολυσακχαρίδωση τύπου IS
- Βλεννοπολυσακχαρίδωση τύπου VI
- Βλεννοπολυσακχαρίδωση τύπου VII
- Σπονδυλοεπιφυσιακές δυσπλασίες (SED)
- Πολλαπλές επιφυσιακές δυσπλασίες (MED)
Πρακτικά, όλοι οι τύποι βραχυσπονδυλικού νανισμού συγχέονται με το σύνδρομο Morquio.
Αχονδροπλασία. Σε αντίθεση με το σύνδρομο Morquio, υπάρχει συνήθως στη γέννηση, αλλά χαρακτηρίζεται από σκελετικές ανωμαλίες τελείως διαφορετικές από τις παρατηρούμενες στην βλεννοπολυσακχαρίδωση τύπου IVA.
MPS I-H. Στην διάρκεια των πρώτων χρόνων της ζωής έχει ακτινολογικές ομοιότητες με το σύνδρομο Morquio, αλλά η διανόηση είναι χαμηλή και η αδρή φυσική εμφάνιση εντυπωσιακά χαρακτηριστική. Θολερότητες του κερατοειδούς και απώλεια της ακοής παρατηρούνται εξίσου στο σύνδρομο Morquio.
Πολλαπλή επιφυσιακή δυσπλασία. Κληρονομείται σύμφωνα με τον επικρατή χαρακτήρα κληρονομικότητας. Μπορεί να υποδύεται σύνδρομο Morquio, αλλά η προσβολή της ΣΣ, εάν υπάρχει, έχει μικρότερη βαρύτητα.
Διαστροφική δυσπλασία (diastrophic dysplasia). Εκδηλώνεται με προοδευτική κυφοσκολίωση, μικρομελία, πλατυποδία, διαπλάτυνση των μεταφύσεων και επιφύσεων των μακρών οστών, μικρό περιορισμό της κινητικότητας των αρθρώσεων και πάχυνση των πτερυγίων των ώτων. Οι εκδηλώσεις αυτές εύκολα διαχωρίζουν το νόσημα αυτό από το σύνδρομο Morquio.
Μετατροπική δυσπλασία (metatropic dysplasia). Τα παιδιά με μετατροπική δυσπλασία αρχικά παρουσιάζουν μακρυκορμικό και αργότερα βραχυκορμικό νανισμό. Χαρακτηρί-ζονται από προοδευτική κυφοσκολίωση και ανισοσπονδυλία, χωρίς υποπλασία του τελευταίου θωρακικού και του πρώτου οσφυικού σπονδύλου.
Συγγενής σπονδυλοεπιφυσιακή δυσπλασία (spondyloepiphyseal dysplasia congenita). Πρόκειται για ανωμαλία του κολλαγόνου τύπου ΙΙ και κληρονομείται σύμφωνα με τον επικρατή χαρακτήρα κληρονομικότητας. Χαρακτηρίζεται από βραχυκορμικό νανισμό και πλατυσπονδυλία, χωρίς όμως προβολή των χεριών και των ποδιών, θολερότητες του κερατοειδούς και αποβολή θειικής κερατάνης από τα ούρα. Μυωπία συχνά είναι σοβαρή.
Σύνδρομο Dyggve-Melchior-Clausen. Εχει αυτοσωμική υπολειπόμενη κληρονομικότητα. Οι πάσχοντες από σύνδρομο Dyggve-Melchior-Clausen έχουν κάποιες ομοιότητες με την νόσο Morquio όσον αφορά τις σκελετικές ανωμαλίες, με τις εξής διαφορές :
- Δεν έχουν θολερότητες του κερατοειδούς,
- Δεν αποβάλλουν KS από τα ούρα
- Δεν έχουν ανεπάρκεια αδαμαντίνης
- Δεν παρουσιάζουν οξύαιχμα μετακάρπια
- Δεν έχουν υποπλασία της οδοντοειδούς απόφυσης
- Δεν έχουν υποπλασία των κατώτερων θωρακικών και οσφυικών σπονδύλων
- Συνήθως έχουν διανοητική καθυστέρηση
- Οι λαγόνιες ακρολοφίες έχουν δαντελωτή παρυφή
Ραχίτιδα
Υποφωσφατασία
Νοσήματα συνδεόμενα με συγγενή δυσπλασία της οδοντοειδούς απόφυσης με υπεξάρθρημα της ατλαντοαξονικής
Συγγενής σπονδυλοεπιφυσιακή δυσπλασία
Σπονδυλομεταφυσιακή δυσπλασία
Σύνδρομο Morquio
Σύνδρομο Aarskog
Σύνδρομο Dyggve-Melchior-Clausen
Υποπλασία χόνδρων – τριχών
Ψευδοαχονδροπλασία
ΘΕΡΑΠΕΙΑ
Συντηρητική θεραπεία
- Μη στεροειδή αντιφλεγμονώδη φάρμακα (ΜΣΑΦ), για τις αρθραλγίες
- Αντιβιοτικά (για τις πνευμονικές λοιμώξεις)
- Συπληρωματικό οξυγόνο (για τις πνευμονικές επιπλοκές ή την αποφρακτική άπνοια ύπνου)
Θεραπείες για ασθενείς με άλλες βλεννοπολυσακκχαριδώσεις
- Ενζυμική θεραπεία αναπλήρωσης (ERT). Προς το παρόν χρησιμοποιείται στη θεραπεία άλλων μεταβολικών νοσημάτων (νόσος Hurler, Maroteaux-Lamy, Gaucher, Fabry, Hunter και Pompe), αλλά όχι της MPS IV
- Γονιδιακή θεραπεία
- Αλλογενική μεταμόσχευση μυελού οστών
- Χειρουργική θεραπεία
- Αυχενική σπονδυλοδεσία, για την πρόληψη του υπεξαρθρήματος της ατλαντοαξονικής
- Μεταμόσχευση κερατοειδούς (για την προοδευτική θολερότητα του κερατοειδούς)
- Μηριαίες οστεοτομίες
- Ολικές αρθροπλαστικές ισχίων ή/και γονάτων
- Χειρουργική διόρθωση βλαισογωνίας
ΠΡΟΓΝΩΣΗ
Στην MPS IV η συχνότητα της νοσηρότητας και θνητότητας σχετίζεται κυρίως με την αστάθεια της ατλαντοαξονικής και την συνακόλουθη αυχενική μυελοπάθεια. Μια μικρή πτώση ή υπερέκταση του αυχένα μπορεί να προκαλέσουν διατομή του νωτιαίου μυελού και τετραπάρεση ή θάνατο. Η αυχενική μυελοπάθεια μπορεί να προκαλέσει δυσλειτουργία του εντέρου και της ουροδόχου κύστης και άπνοια.
Η αποφρακτική άπνοια ύπνου μπορεί να οδηγήσει σε μακροχρόνια υποξία, πνευμονική υπέρταση, ακόμα και θάνατο.
Οι ασθενείς με MPS IV, λόγω της προοδευτικής παραμόρφωσης του κορμού και της ακινησίας, έχουν αυξημένη προδιάθεση για πνευμονικές λοιμώξεις. Μπορεί ακόμα να παρουσιάσουν πρώιμη στεφανιαία καρδιοπάθεια και πάχυνση των καρδιακών βαλβίδων (αορτής και μιτροειδούς) με συνακόλουθη καρδιακή δυσλειτουργία.
Οι ασθενείς με ήπιες εκδηλώσεις συνδρόμου Morquio, ανεξαρτήτως τύπου, μπορεί να επιβιώσουν μέχρι το 70ό έτος της ηλικίας. Οι ασθενείς με σοβαρές εκδηλώσεις, κυρίως σχετιζόμενες με αυχενική αστάθεια και πνευμονική ανεπάρκεια, συχνά δεν επιβιώνουν πέραν της 2ης ή 3ης δεκαετίας της ζωής.
5. ΒΛΕΝΝΟΠΟΛΥΣΑΚΧΑΡΙΔΩΣΗ ΤΥΠΟΥ VI (Σύνδρομο Maroteaux-Lamy) (Mucopolysaccharidosis VI; MPS VI; Maroteaux-Lamy syndrome)
ΧΑΡΑΚΤΗΡΙΣΤΙΚΑ ΝΟΣΟΥ
Η βλεννοπολυσακχαρίδωση τύπου 6 (σύνδρομο Maroteaux-Lamy) (McKusick 253200) είναι νόσημα θησαυρισμού μεταβιβαζόμενο σύμφωνα με το αυτοσωμικό υπολειπόμενο πρότυπο κληρονομικότητας. Κλινικά χαρακτηρίζεται από :
- βραχυσωμία,
- οργανομεγαλία,
- σκελετικές ανωμαλίες,
- θόλωση του κερατοειδούς,
- διόγκωση της γλώσσας,
- τράχυνση των χαρακτηριστικών του προσώπου,
- βαρηκοΐα,
- χρόνιες αναπνευστικές λοιμώξεις,
- δυσκαμψία των αρθρώσεων και
- καρδιακή ανεπάρκεια λόγω προσβολής των βαλβίδων.
Οι ασθενείς με MPS VI αρχικά συνήθως αναπτύσσονται φυσιολογικά, αλλά αργότερα παρουσιάζουν συχνές αναπνευστικές λοιμώξεις ή μέση ωτίτιδα, βουβωνοκήλες ή ομφαλοκήλες, θολερότητες του κερατοειδούς, ηπατοσπληνομεγαλία, τράχυνση των χαρακτηριστικών του προσώπου και ανωμαλίες των αρθρώσεων ή της σπονδυλικής στήλης.
ΤΥΠΟΙ ΒΛΕΝΝΟΠΟΛΥΣΑΚΚΧΑΡΙΔΩΣΗΣ VI (MPS VI)
H MPS VI διακρίνεται σε 3 τύπους :
- Ηπιος τύπος (Paterson DE et al, 1982; Poser C et al, 1983),
- Ενδιάμεσος τύπος και
- Βαρύς τύπος (Spranger J et al, 1970; Spranger J, 1972; Rampini S et al, 1986).
Μπορεί ακόμα να υπάρχει ένας πολύ ήπιος τύπος (Saul RA et al, 1984).
Τα παιδιά με ήπιο τύπο MPS VI αναπτύσσονται σχετικά καλά μέχρι το 6ο έτος της ηλικίας, οπότε αναπτύσσουν βραχυσωμία, θολερότητες του κερατοειδούς, παραμορφώσεις της σπονδυλικής στήλης, προσβολή των ισχίων παρόμοια με νόσο Legg-Calve-Perthes και στένωση της αορτής. Οι ασθενείς συνήθως επιβιώνουν μέχρι την ενήλικη ζωή (Paterson DE et al, 1982).
Στους ασθενείς με βαρύ τύπο οι μορφολογικές μεταβολές παρουσιάζονται στη πρώιμη παιδική ηλικία και η νόσος εξελίσσεται ταχύτερα σε σοβαρή αναπηρική κατάσταση, με μεγάλη βραχυσωμία, μυοσκελετικές ανωμαλίες, τράχυνση των χαρακτηριστικών του προσώπου, υπερέκταση της κεφαλής, σημαντική εξασθένηση της ακοής και καρδιακές ανωμαλίες, οι οποίες συχνά οδηγούν στο θάνατο στην εφηβική ηλικία.
ΕΠΙΔΗΜΙΟΛΟΓΙΑ
Συχνότητα. Η συχνότητα της MPS VI διαφέρει στους διάφορους πληθυσμούς και γεωγραφικές περιοχές, κυμαινόμενη από 1/238.000 έως 1.298.000 (Lowry RB, 1990; Nelson J, 1997; Poorthuis BJ et al, 1999; Meikle PJ et al, 1999; Pinto R et al, 2004) (ΠΙΝΑΚΑΣ 5).
Στην Αυστραλία, στο διάστημα των ετών 1980-1996, η συχνότητα της MPS VI είχε υπολογισθεί σε 1 περίπτωση/248.000 γεννήσεις (Meikle PJ et al, 1999).
Στη Γερμανία και την Βόρεια Πορτογαλία, υπολογίζεται σε 1/432. 610 και 1/238.095 γεννήσεις, αντίστοιχα (Pinto R et al, 2004).
Με βάση την συνδυασμένη ανάλυση των πλέον πρόσφατων μελετων, η μέση παγκόσμια συχνότητα της MPS VI υπολογίζεται σε 1/340,000. Περίπου 1.100 άτομα μπορεί να πάσχουν παγκοσμίως από MPS VI, αν και πολύ λιγότεροι έχουν διαγνωσθεί μέχρι σήμερα (Swiedler SJ et al, 2005).
Φυλή. Η MPS VI δεν έχει φυλετική προτίμηση, αν και απαντάται σε αυξημένη συχνότητα σε άτομα Τουρκικής καταγωγής που διαμένουν στη Γερμανία.
Φύλο. Η MPS VI απαντάται εξίσου και στα 2 φύλα.
Ηλικία. Η MPS VI εκδηλώνεται τυπικά στην πρώιμη παιδική ηλικία.
ΠΙΝΑΚΑΣ 5.
Συχνότητα MPS VI (σύμφωνα με τις μελέτες)
Γεωγραφική περιοχή (ζώντα βρέφη) Ετη Συχνότητα MPS VI
Βόρεια Πορτογαλία 1982–2001 1/238,000
Αυστραλία 1980–1996 1/248,000
Ολλανδία 1970–1990 1/657,000
Βόρεια Ιρλανδία 1958–1985 <1/840,000
Βρεττανική Κολομβία, Καναδάς 1952–1986 1/1,298,000
ΑΙΤΙΟΛΟΓΙΑ
Το σύνδρομο Maroteaux-Lamy οφείλεται σε ανεπάρκεια της Ν- ακετυλ-γαλακτοζαμινο-IV-σουλφατάσης (αρυλσουλφατάση Β) (Litjens T et al, 1996). Η αρυλσουλφατάση Β είναι ένα λυσοσωμικό ένζυμο, το οποίο καταλύει την αποδόμηση της θειικής δερματάνης. Το υπεύθυνο γονίδιο εντοπίζεται στη χρωμοσωμική περιοχή 5q11-q13.
Η ανεπάρκεια της αρυλσουλφατάσης Β οδηγεί σε αναστολή της αποδόμησης των βλεννοπολυσακχαριδών και εναπόθεση θειικής δερματάνης στο σκελετό, τις καρδιακές βαλβίδες, το ήπαρ, τον σπλήνα και τον κερατοειδή, δίνοντας γένεση στις πολλές και ποικίλες εκδηλώσεις της νόσου. Η νόσος μεταβιβάζεται σύμφωνα με το αυτοσωμικό υπολειπόμενο πρότυπο κληρονομικότητας.
ΚΛΙΝΙΚΗ ΕΙΚΟΝΑ
Οι κλινικές εκδηλώσεις του συνδρόμου Maroteaux-Lamy είναι ήπιες έως σοβαρές. Αρχικά, τα παιδιά με σύνδρομο Maroteaux-Lamy γεννιούνται φυσιολογικά. Κατά το 2ο όμως έτος της ηλικίας, παρουσιάζουν βραχυσωμία και αργότερα οσφυική κύφωση, προβολή του στέρνου και άλλες ανωμαλίες του σκελετού και των μαλακών μορίων, τράχυνση των χαρακτηριστικών του προσώπου, ηπατοσπληνομεγαλία, θόλωση του κερατοειδούς και συγκάμψεις των αρθρώσεων.
Οι πάσχοντες τυπικά έχουν φυσιολογική νοημοσύνη, αλλά αργότερα μπορεί να εμφανίσουν εξασθένηση της αναγνώρισης, πιθανώς σε συνδυασμό με υδροκεφαλία, και συνήθως καταλήγουν κακώς στην εφηβική ή στη πρώιμη ενήλικη ζωή, συνήθως από καρδιοαναπνευστική ανεπάρκεια.
Διαταραχές ακοής - όρασης. Οι ασθενείς με MPS VI συχνά παρουσιάζουν διαταραχές της ακοής, τόσο νευροαισθητήριες, όσο και αγωγιμότητας και, μερικοί, σημαντικές θολερότητες του κερατοειδούς (Neufeld EF and Muenzer J, 1995), όπως και γλαύκωμα (Paterson DE et al, 1982).
Μυοσκελετικές ανωμαλίες. Το ύψος των ενηλίκων με MPS VI ανέρχεται συνήθως σε 110–140 cm, αλλά στους ασθενείς με τον ήπιο τύπο της νόσου μπορεί να φθάσει τα 168 cm (Pilz H et al, 1979).
Ο θώρακας παραμορφώνεται και το στέρνο προβάλλει. Μετά το 1ο έτος της ηλικίας αναπτύσσονται πολλαπλές αρθρικές συγκάμψεις και γαμψοδακτυλία. Τα γόνατα συχνά παρουσιάζουν βλαισογωνία και η ΣΣ, οσφυική κύφωση.
Γαστρεντερικές ανωμαλίες. Ηπατομεγαλία έχουν σχεδόν όλοι οι ασθενείς με βαρύ MPS VI. Ο σπλήνας διογκώνεται στο 50% περίπου των περιπτώσεων. Κήλες είναι συχνές σε ασθενείς με τον βαρύ τύπο της νόσου.
Καρδιακές επιπλοκές. Συχνά ακόμα οι ασθενείς με MPS IV έχουν μυοκαρδιοπάθεια (η οποία μπορεί να είναι η πρώτη εκδήλωση της νόσου), παλινδρόμηση της μιτροειδούς, ενδοκαρδιακή ινοελάστωση και στένωση της αορτής, των στεφανιαίων και άλλων αρτηριών (Schieken RN et al, 1975; Fong LV et al, 1987; Hayflick S et al, 1992; Marwick TH et al, 1992; Tan CTT et al, 1992).
Νευρολογικές διαταραχές. Η διανόηση είναι σχεδόν πάντα φυσιολογική, με σπάνιες εξαιρέσεις (Taylor HR et al, 1978; Vestermark S et al, 1987).
Οι νευρολογικές διαταραχές περιλαμβάνουν υδροκέφαλο, συμπίεση περιφερικών νεύρων (π.χ. σύνδρομο καρπιαίου σωλήνα) ή υποπλασία της οδοντοειδούς απόφυσης συνδεόμενη με αστάθεια της ατλαντοαξονικής.
Μυελοπάθεια και ριζοπάθεια έχουν επίσης αναφερθεί (Goldberg MF et al, 1970; Peterson DI et al, 1975; Rampini S et al, 1986).
Κρανιοπροσωπικές ανωμαλίες. Τα παιδιά με MPS VI μπορεί να γεννηθούν με προπέτεια του μετώπου. Το προσωπείο τους παρουσιάζεται περί το 6ο έτος της ηλικίας και, σε μερικές περιπτώσεις, ενωρίτερα (Spranger J et al, 1970; Van Biervliet JP et al, 1977). Είναι παρόμοιο με του συνδρόμου Hurler, χαρακτηριζόμενο από υπερτελορισμό, συμπίεση της ρινικής γέφυρας, κρανιομεγαλία, «γεμάτα» μάγουλα και χείλη, σχετικά πλατειά γνάθο, πυκνές βλεφαρίδες και πυκνά μαλλιά.
Στόμα - γνάθος. Η γλώσσα διογκώνεται και τα μεσοδόντια διαστήματα είναι αραιά. Η ανατολή της πρωτογενούς και μόνιμης οδοντοφυίας αναστέλλεται (Nakamura T et al, 1992; Smith KS et al, 1995). Οι κόνδυλοι της κάτω γνάθου είναι υποπλαστικοί. Συχνά αναπτύσσεται σοβαρή στένωση της τραχείας (Rampini S et al, 1987).
ΚΛΙΝΙΚΕΣ ΕΚΔΗΛΩΣΕΙΣ (ΣΥΝΟΠΤΙΚΑ)
Κεφαλή – πρόσωπο-στόμα
- Αποπλάτυνση γνάθου
- Διόγκωση παρειών
- Διόγκωση της γλώσσας
- Καθυστέρηση οδοντοφυίας
- Μακροκεφαλία
- Πάχυνση των χειλέων
- Πάχυνση μυκτήρων
- Προβολή μετώπου
- Συμπίεση ρινικής γέφυρας
- Τράχυνση των χαρακτηριστικών του προσώπου
- Υπερτελορισμός
Μυοσκελετικό
- Γαμψοδακτυλία
- Γωνίωση σπονδύλων
- Βλαισογωνία
- Κύφωση
- Νέκρωση επιφύσεων μηριαίας κεφαλής
- Συμπίεση νωτιαίου μυελού
- Συγκάμψεις των αρθρώσεων με περιορισμό της κινητικότητας (Saul RA et al, 1984)
Αισθητήρια όργανα
- Βαρηκοΐα
- Γλαύκωμα (Cantor LB et al, 1989)
- Εξασθένηση όρασης
- Θολερότητες κερατοειδούς (Saul RA et al, 1984)
- Οίδημα οπτικής θηλής
Θώρακας -κοιλιά
- Παραμόρφωση με προβολή του στέρνου και αποπλάτυνση των πλευρών
- Ηπατομεγαλία/σπληνομεγαλία
- Ομφαλοκήλη ή/και βουβωνοκήλη
- Εξαρτήματα δέρματος : Πάχυνση των μαλλιών και των βλεφαρίδων
Καρδιαγγειακό : Στένωση αορτής και μιτροειδούς (Wilson CS et al, 1980; Tan CT et al, 1992).
Κεντρικό – περιφερικό νευρικό σύστημα
- Απνοια ύπνου
- Αυχενική μυελοπάθεια λόγω υπεξαρθρήματος της ατλαντοαξονικής ή πάχυνσης της σκληράς μήνιγγας (Young R et al, 1980; Poser CM et al, 1983), η οποία μπορεί να οδηγήσει σε αδυναμία των μυών των άνω – κάτω άκρων, παράλυση ή παραπληγία
- Διανοητική καθυστέρηση (Vestermaark S et al, 1987)
- Συμπίεση περιφερικών νεύρων (π.χ. σύνδρομο καρπιαίου σωλήνα)
- Υδροκέφαλος με αυξημένη ενδοκρανιακή πίεση λόγω προσβολής των μηνίγγων (Schwartz GP and Cohen EJ, 1998)
Αναπνευστικό
- Περιοριστική πνευμονοπάθεια, οφειλόμενη σε περιορισμό της έκπτυξης του θώρακα, δευτεροπαθώς σε κυφοσκολίωση
- Στένωση της τραχείας
ΕΡΓΑΣΤΗΡΙΑΚΑ ΕΥΡΗΜΑΤΑ
Τα κοκκιοκύτταρα και τα μονοκύτταρα και μεγάλο ποσοστό λεμφοκυττάρων του περιφερικού αίματος παρουσιάζουν άφθονα τραχέα, πυκνά έγκλειστα. Παρόμοια, ο μυελός των οστών παρουσιάζει τραχέα έγκλειστα στα δικτυοιστιοκύτταρα, κοκκιοκύτταρα και στις πρόδρομες μορφές τους (Markesbery WR et al, 1980).
Στο ηλεκτρονικό μικροσκόπιο παρατηρούνται αρκετά κενοτόπια στα κύτταρα του εγκεφάλου, του ήπατος, των πνευμόνων και του δέρματος (Levy LA et al, 1980; Libert J, 1980).
Οι ασθενείς με MPS VI αποβάλλουν μεγάλες ποσότητες DS από τα ούρα, αλλά τα επίπεδά τους μειώνονται με την πάροδο της ηλικίας.
ΑΚΤΙΝΟΛΟΓΙΚΑ ΕΥΡΗΜΑΤΑ
- Αγκιστροειδής παραμόρφωση ή αμβλεία πρόσθια υποπλασία των Ο1 και Ο2 σπονδύλων
- Αμφίκυρτη διαμόρφωση τελικών σπονδυλικών πλακών
- Βράχυνση, διαπλάτυνση και όξυνση των μετακαρπίων
- Βλαισό ισχίο
- Διαπλάτυνση λαγόνιων ακρολοφιών
- Διόγκωση τουρκικού εφιππίου
- Δυσπλασία επίφυσης μηριαίας κεφαλής
- Επιμήκυνση του αυχένα του μηριαίου σε θέση βλαισότητας
- Κατάτμηση των επιφύσεων
- Παραμόρφωση των μακρών σωληνωδών οστών
- Υποπλασία οδοντοειδούς απόφυσης και υπεξάρθρημα της ατλαντοαξονικής
- Υποπλασία κοτύλης-λαγονίου
ΔΙΑΦΟΡΙΚΗ ΔΙΑΓΝΩΣΗ
- Βλεννοπολυσακκχαρίδωση τύπου II
- Βλεννοπολυσακκχαρίδωση τύπου VII
- Βλεννοπολυσακκχαρίδωση τύπου I H/S
- Βλεννοπολυσακκχαρίδωση τύπου IH
- Βλεννοπολυσακκχαρίδωση τύπου IS
- Πολλαπλή ανεπάρκεια σουλφατάσης
- Βλεννολιπιδώσεις
ΘΕΡΑΠΕΙΑ
1. Ενζυμική θεραπεία αναπλήρωσης
Η ανασυνδυασμένη ποικιλία DNA της N-ακετυλγαλακτοζαμινο- 4-σουλφατάσης μπορεί να βελτιώσει τις φυσικές ικανότητες των ασθενών με MPS VI. Η ενζυμική θεραπεία αναπλήρωσης με galsulfase (Naglazyme) βελτιώνει την βάδιση και την ικανότητα ανάβασης κλίμακας και μειώνει τα επίπεδα των γλυκοζαμινογλυκανών στα ούρα σε ασθενείς με MPS VI
Δόσεις :
- Ενήλικες : 1 mg/kg ενδοφλεβίως σε διάστημα 4 ωρών qwk
Παιδιά :
- < 5 ετών : Δεν έχει προσδιορισθεί
- ≥ 5 ετών : Δόσεις ενηλίκων
2. ΧΕΙΡΟΥΡΓΙΚΗ ΘΕΡΑΠΕΙΑ
- Αμυγδαλεκτομή και αδενοειδεκτομή (σε ασθενείς με αποφρακτική πνευμονοπάθεια)
- Αντικατάσταση καρδιακών βαλβίδων (σε ασθενείς με βαλβιδοπάθειες)
- Μεταμόσχευση κερατοειδούς (για τις θολερότητες του κερατοειδούς)
- Ολική αρθροπλαστική ισχίου (για την δυσπλασία των ισχίων)
- Τραχειοστομία, σε περιπτώσεις σοβαρής απόφραξης των αναπνευστικών οδών και υπαερισμού
- Χειρουργική αποσυμπίεση του καρπιαίου σωλήνα (σε περιπτώσεις συνδρόμου καρπιαίου σωλήνα)
- Κοιλιοπεριτοναϊκή παράκαμψη (για τον υδροκέφαλο ή την αυξημένη ενδοκρανιακή πίεση)
- Χειρουργική αποσυμπίεση νωτιαίου μυελού ή σταθεροποίηση ατλαντοαξονικής συμβολής
ΠΡΟΓΝΩΣΗ
Η MPS VI χαρακτηρίζεται από σημαντική νοσηρότητα και πρόωρη θνητότητα, λόγω της προοδευτικής άθροισης των γλυκοζαμινογλυκανών. Η ηλικία θανάτου ποικίλλει. Οι ασθενείς με ήπιους τύπους της νόσου μπορεί να επιβιώσουν μακροχρόνια.
ΒΛΕΝΝΟΠΟΛΥΣΑΚΧΑΡΙΔΩΣΗ ΤΥΠΟΥ VII (σύνδρομο Sly) (Mucopolysaccharidosis VII; MPS VII; Sly syndrome)
ΧΑΡΑΚΤΗΡΙΣΤΙΚΑ ΝΟΣΟΥ
Το σύνδρομο Sly (MPS-VII) (McKusick 253220) είναι πολύ σπάνιο λυσοσωμικό νόσημα θησαυρισμού. Φαινοτυπικά χαρακτηρίζεται κυρίως από βραχυσωμία, ηπατοσπληνομεγαλία, προοδευτική πολλαπλή δυσόστωση και ήπια προοδευτική διανοητική καθυστέρηση μετά το 2ο έτος της ηλικίας.
ΕΠΙΔΗΜΙΟΛΟΓΙΑ
Συχνότητα. Η MPS VII είναι εξαιρετικά σπάνια. Μερικές μόνο περιπτώσεις έχουν αναφερθεί.
Φύλο. Η MPS VII απαντάται εξίσου και στα 2 φύλα.
Ηλικία. Το σύνδρομο Sly είναι κλινικά εμφανές στη γέννηση. Οι ηπιότεροι τύποι παρουσιάζονται συνήθως αργότερα.
ΑΙΤΙΟΛΟΓΙΑ
Το σύνδρομο Sly είναι οικογενές, μεταδιδόμενο σύμφωνα με το αυτοσωμικό υπολειπόμενο πρότυπο κληρονομικότητας, και οφείλεται σε μεταλλάξεις του γονιδίου το οποίο κωδικοποιεί την β-γλυκουρονιδάση (GUSB). Το γονίδιο αυτό έχει χαρτογραφηθεί στο μακρό σκέλος του χρωμοσώματος 7 (7q11. 21-7q11.22) (Knowles ΒΒ et al, 1977). Το GUSP έχει μήκος 21-kb και περιέχει 12 εξόνια. Περισσότερες από 45 διαφορετικές μεταλλάξεις έχουν πιστοποιηθεί, εκ των οποίων περίπου 90% είναι σημειακές.
Η ανεπάρκεια της β-γλυκουρονιδάσης οδηγεί σε άθροιση θειικής δερματάνης, θειικής ηπαράνης και θειικής χονδροϊτίνης σε διάφορους ιστούς και όργανα, η οποία συμβάλλει στις διάφορες μορφολογικές ανωμαλίες που χαρακτηρίζουν την νόσο.
ΚΛΙΝΙΚΗ ΕΙΚΟΝΑ
Οι εκδηλώσεις της MPS VII έχουν εξαιρετική ποικιλομορφία, αλλά γενικά έχουν μεγάλες ομοιότητες με τις εκδηλώσεις του συνδρόμου Hurler και των άλλων βλεννοπολυσακχαριδώσεων (Sly WS et al, 1973; Lee JE et al, 1985; Pizzutillo PD et al, 1989). Η MPS VII κλινικά χαρακτηρίζεται από :
- βραχυσωμία λόγω αναστολής της ανάπτυξης,
- χαρακτηριστικές ακτινολογικές αλλοιώσεις και
- ήπια διανοητική καθυστέρηση (Sly WS et al, 1973; Vervoort R et al, 1996; Stone DL and Sidransky E, 1999).
Συχνά ακόμα εκδηλώνεται με μη αυτοάνοσο εμβρυικό ύδρωπα, ο οποίος όμως συνήθως δεν αποδίδεται στη νόσο, δοθέντος ότι οι πάσχοντες κατά κανόνα καταλήγουν κακώς πριν διαγνωσθούν (de Kremer RD et al, 1992)
Άλλες εκδηλώσεις περιλαμβάνουν :
- δυσμορφία,
- κήλες,
- ηπατοσπληνομεγαλία,
- πλατυποδία,
- δυσόστωση,
- σοβαρή υποτονία και
- νευρολογικές διαταραχές, οι οποίες τελικά οδηγούν σε σοβαρή διανοητική καθυστέρηση
Στις ηπιότερες περιπτώσεις οι ασθενείς επιβιώνουν μέχρι την ενηλικίωση και συχνά αναπτύσσουν οστεοαρθρίτιδα. Οι πολύ ήπιες περιπτώσεις ανακαλύπτονται στην εφηβική ηλικία ή στην ενηλικίωση λόγω θωρακικής σκολίωσης.
ΤΥΠΟΙ MPS VII
Η MPS VII μπορεί να παρουσιασθεί κλινικά με τους εξής τύπους :
1. Ένα σοβαρό και πρώιμης έναρξης τύπο, ο οποίος παρουσιάζεται στη διάρκεια των 4 πρώτων ετών της ζωής. Ο τύπος αυτός περιλαμβάνει τον προνεογνικό τύπο, ο οποίος εκδηλώνεται με μη άνοσο εμβρυικό ύδρωπα, και σοβαρούς νεογνικούς τύπους οι οποίοι παρουσιάζονται με νεογνική χολόσταση και ηπατοσπληνομεγαλία, και
2. Εναν όψιμης έναρξης τύπο, ο οποίος απαντάται συνήθως μετά το 4ο έτος της ηλικίας και χαρακτηρίζεται από ηπιότερα συμπτώματα.
ΚΛΙΝΙΚΕΣ ΕΚΔΗΛΩΣΕΙΣ
Νευρολογικές διαταραχές. Τα 2 πρώτα χρόνια της ζωής, οι βαρέως πάσχοντες από MPS VII παρουσιάζουν σοβαρή αναστολή της ανάπτυξης, υποτονία και νευρολογικές ανωμαλίες, οι οποίες τελικά οδηγούν σε διανοητική καθυστέρηση. Η διανοητική καθυστέρηση είναι συνήθως μέτρια, μη προοδευτική και εντονότερη στον λόγο και την γλωσσική ανάπτυξη. Στις βαρύτερες περιπτώσεις, η MPS VII μπορεί να παρουσιασθεί με μη αυτοάνοσο εμβρυικό ύδρωπα (Nelson A et al, 1982; Kagia MJ et al, 1992; Stagenberg M et al, 1992; Vervoort R et al, 1996).
Διαταραχές ακοής. Οι ασθενείς με σύνδρομο Sly μπορεί να εμφανίσουν απώλεια της ακοής.
Διαταραχές όρασης. Μετά το 1ο έτος της ηλικίας οι ασθενείς με MPS VII μπορεί να παρουσιάσουν θολερότητες του κερατοειδούς και κολόβωμα της ίριδας.
Σκελετικές ανωμαλίες. Η περιφερική δυσόστωση συνδέεται με τους βαρύτερους τύπους MPS II. Οι σκελετικές ανωμαλίες περιλαμβάνουν παρεκτόπιση των ισχίων, συγκάμψεις των αρθρώσεων, κυφοσκολίωση και πλατύ θωρακικό κλωβό. Κωνοειδής θώρακας ή «θώρακας των υποδηματοποιών» και θωρακοοσφυικός ύβος, ο οποίος ήδη υπάρχει από την βρεφική ηλικία, επιδεινώνεται με την πάροδο της ηλικίας (de Kremer RD et al, 1992).
Κρανιοπροσωπικές ανωμαλίες. Δυσμορφικές εκδηλώσεις είναι συχνές και χαρακτηρίζονται από τράχυνση των χαρακτηριστικών του προσώπου, μακροκεφαλία, προβολή του μετώπου, πρόωρη σύγκλειση των τοξοειδών λαβδοειδών ραφών και βραχυλαιμία.
Γαστρεντερικές διαταραχές. Χαρακτηριστική εκδήλωση της MPS VII είναι ηπατομεγαλία ή ηπατοσπληνομεγαλία.
Άλλες εκδηλώσεις :
- Γαστρεντερικές διαταραχές και ασκίτης
- Βουβωνοκήλες και ομφαλοκήλες
- Αγγειακές ανωμαλίες (σπάνια)
- Βαλβιδοπάθειες και παλινδρόμηση αορτής
- Λεμφοίδημα/οίδημα
- Νεογνικός ίκτερος
- Πνευμονικές ανωμαλίες
- Υπερτρίχωση
- Υποτροπιάζουσες λοιμώξεις του ανώτερου αναπνευστικού
ΚΛΙΝΙΚΕΣ ΕΚΔΗΛΩΣΕΙΣ (ΣΥΝΟΠΤΙΚΑ)
Κρανίο και αυχένας
- Βραχυλαιμία
- Μακροκεφαλία
- Πρόσθια κλίση μυκτήρων
- Προβολή φατνιακών αποφύσεων
- Προβολή του μετώπου
- Συμπίεση ρινικής γέφυρας
- Τράχυνση των χαρακτηριστικών του προσώπου (όπως στο σύνδρομο Hurler)
- Υπερωϊοσχιστία
Οφθαλμοί
- Θόλωση κερατοειδούς
- Κολοβώματα ίριδας
- Υπερτελορισμός
Θώρακας -πλευρές
- Ανωμαλία υπεζωκότα/υδροθώρακας
- Τροπιδοειδής ή χωνοειδής θώρακας
Κοιλιά
- Ασκίτης
- Βουβωνοκήλες/ομφαλοκήλες
- Ηπατομεγαλία
- Προβολή κοιλιάς
- Σπληνομεγαλία
Οστά και αρθρώσεις
- Ασηπτη νέκρωση μηριαίας κεφαλής
- Εξάρθρημα ισχίου
- Πολλαπλή δυσόστωση
- Ραιβοκοιλοποδία
- Κύφωση ή σκολίωση
Άλλες
- Αγγειακές ανωμαλίες
- Αναστολή σωματικής, κινητικής και διανοητικής ανάπτυξης
- Οίδημα/λεμφοίδημα
- Υποτροπιάζουσες αναπνευστικές λοιμώξεις
ΕΡΓΑΣΤΗΡΙΑΚΑ ΕΥΡΗΜΑΤΑ
Βλεννοπολυσακκχαρουρία ή αύξηση των επιπέδων των γλυκοζαμινογλυκανών στα ούρα (είτε θειικής χονδροϊτίνης μόνης της ή θειικής δερματάνης, είτε θειικής ηπαράνης σε συνδυασμό με θειική χονδροϊτίνη)
ΙΣΤΟΛΟΓΙΚΑ ΕΥΡΗΜΑΤΑ
- Τραχειά μεταχρωματικά έγκλειστα σε περιφερικά κοκκιοκύτταρα
- Πρόδρομοι των κοκκιοκυττάρων στον μυελό των οστών και σε καλλιεργημένους ινοβλάστες
- Καθαρά κενοτόπια και κοκκιώδη έγκλειστα σε όλα σχεδόν τα κοκκιοκύτταρα και μονοπύρηνα κύτταρα (στο ηλεκτρονικό μικροσκόπιο) (Peterson L et al, 1982)
ΑΚΤΙΝΟΛΟΓΙΚΑ ΕΥΡΗΜΑΤΑ
Χέρια
- Εγγυς όξυνση του 2ου-5ου μετακαρπίου
- Πλευρές
- Πλευρές σχήματος «κουπιού»
Ισχία-λεκάνη
- Ασηπτη νέκρωση μηριαίας κεφαλής
- Γενικευμένη οστεοσκλήρυνση
- Παρεκτόπιση των ισχίων
- Υπανάπτυξη των βασικών τμημάτων των λαγονίων
Κρανίο
- Παραμόρφωση των οδόντων και πτωχή ανάπτυξη των μαστοειδών αποφύσεων και των παραρρινίων κόλπων
- Πρόωρη σύγκλειση των οβελιαίων λαβδοειδών ραφών
- Τουρκικό εφίππιο σχήματος «J»
Σπονδυλική στήλη
- «Αγκιστροειδείς» παραμορφώσεις των κατώτερων θωρακικών και των ανώτερων οσφυικών σπονδύλων
- Θωρακοοσφυική κυφοσκολίωση
- Πλατυσπονδυλία
- Προέχουσα πρόσθια αγγειακή εντομή των σπονδυλικών σωμάτων και διάχυτη πύκνωση
- Υποπλασία οδοντοειδούς απόφυσης
Σωληνώδη οστά
- Βράχυνση των σωληνωδών οστών
- Διαταραχή της αρχιτεκτονικής των σωληνωδών οστών με επιμήκεις ή κάθετες γραμμώσεις (παρόμοια με ραχίτιδα)
Αλλα. Περιοστίτιδα και κατάγματα
ΔΙΑΓΝΩΣΗ
Η οριστική διάγνωση της MPS VII μπορεί να γίνει με την ανεύρεση της ανεπάρκειας της β-γλυκουρονιδάσης στον ορό, σε λευκά αιμοσφαίρια ή σε καλλιεργημένους ινοβλάστες του δέρματος. Μπορεί ακόμα να γίνει μοριακή ανάλυση του γονιδίου GUSB.
Η προνεογνική διάγνωση της ενζυμικής ανεπάρκειας μπορεί να γίνει με ενζυμικές εξετάσεις του αμνιακού υγρού και μοριακή ανάλυση κυττάρων του αμνιακού υγρού και των χοριακών λαχνών (Maire I et al, 1980).
ΔΙΑΦΟΡΙΚΗ ΔΙΑΓΝΩΣΗ
- Βλεννοπολυσακκχαρίδωση τύπου IH
- Βλεννοπολυσακκχαρίδωση τύπου II
- Βλεννολιπιδώσεις
- Ολιγοσακκχαρίδωση
ΘΕΡΑΠΕΙΑ
- Αντιμετώπιση αναπνευστικών και καρδιαγγειακών επιπλοκών
- Βελτίωση όρασης και ακοής
- Θεραπεία επικοινωνούντα υδροκέφαλου
- Βελτίωση γαστρεντερικών συμπτωμάτων
- Χειρουργική διόρθωση αρθρικών συγκάμψεων και παραμορφώσεων χεριών και ποδιών
- Μεταμόσχευση κερατοειδούς, εάν η όραση έχει εξασθενήσει σοβαρά
ΠΡΟΓΝΩΣΗ
Οι μετανεογνικοί τύποι MPS VII έχουν κακή πρόγνωση και συνήθως καταλήγουν στο θάνατο in utero. Οι νεογνικοί και παιδικοί τύποι έχουν πολύ περιορισμένο προσδόκιμο επιβίωσης, ενώ οι πάσχοντες από ηπιότερους τύπους μπορεί να επιβιώσουν μέχρι το 19ο-20ο έτος της ηλικίας.
Η διάρκεια της επιβίωσης μπορεί να επηρεάζεται από τις λοιμώξεις του ανώτερου αναπνευστικού και τις νευροεκφυλιστικές και γαστρεντερικές επιπλοκές.

Θέματα
Συλλογή Διαφανειών
 JPEG.jpg)
 JPEG.jpg)
 Πόδια JPEG.jpg)
 Γόνατα JPEG.jpg)
 Χέρια JPEG.jpg)
Συλλογή Φωτογραφιών

 Image Collection.jpg)
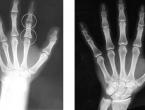

.jpg)
.jpg)
.jpg)

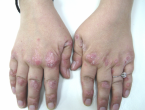
 KONSTANTINA Πρόσωπο (1).jpg)

.jpg)
.jpg)
.jpg)
 Φωτο + αα.jpg)



.jpg)

 JPEG.jpg)
 Αγκώνας JPEG.jpg)
 Χέρια JPEG.jpg)
_0.jpg)
_0.jpg)
 Μαλτέζος Πρόσωπο (4).jpg)
 KONSTANTINA Χέρια (3).jpg)
.jpg)
.jpg)
 JPEG.jpg)
 JPEG.jpg)
 JPEG.jpg)
 JPEG.jpg)
 JPEG.jpg)
 JPEG.jpg)
 JPEG.jpg)
 JPEG Γόνατα.jpg)
 JPEG Αγκώνες (2).jpg)
.jpg)
 Σημείο αντίχειρα.jpg)
 άγνωστης αιτιολογίας.jpg)
 Φωτο (2) Παθητική ανάταξη (1).jpg)

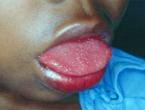
.jpg)

.jpg)
.jpg)
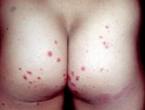
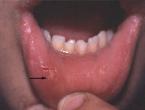
.jpg)

.jpg)
.jpg)
 Χέρια Φωτο.jpg)
 JPEG.jpg)
 Χέρια Φωτο.jpg)
.jpg)
.jpg)
 Σημείο Gottron (1).jpg)
 Σημείο Gottron (2).jpg)
 Υποδόριες ασβεστώσεις (1).jpg)
 Υποδόριες ασβεστώσεις (2).jpg)
 Χέρια JPEG.jpg)
 Χέρια JPEG.jpg)
 Χέρια JPEG.jpg)
 Χέρια JPEG.jpg)
 Χέρια JPEG.jpg)
 Χέρια JPEG.jpg)
 Χέρια JPEG.jpg)
 Χέρια JPEG.jpg)
 Χέρια JPEG.jpg)
 Χέρια JPEG.jpg)
 Χέρια JPEG.jpg)
 Χέρια JPEG.jpg)
 Εξάνθημα JPEG.jpg)
 JPG.jpg)
 JPG.jpg)
 JPG_0.jpg)
 JPG_0.jpg)
 JPG.jpg)
 Οσφύς JPEG.jpg)
 Οσφύς JPEG.jpg)
 Φωτο.jpg)
 Φωτο.jpg)
 Βραχυδακτυλία JPEG.jpg)


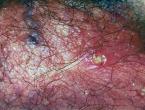
_2.jpg)
_0.jpg)
 Αρθρίτιδα χεριών.jpg)
 Δερματικό εξάνθημα.jpg)
.jpg)
_0.jpg)
.jpg)
 Υποδόριες ασβεστώσεις (1)_0.jpg)
 Υποδόριες ασβεστώσεις (2)_0.jpg)
_0.jpg)
 Χέρια Φωτο_0.jpg)
 Χέρια Φωτο_0.jpg)
 Χέρια JPEG.jpg)
 Aρθροπάθεια.jpg)
 Αγγειοκερατώματα.jpg)
_1.jpg)
.jpg)
.jpg)
_2.jpg)
 (2).jpg)
_1.jpg)
.jpg)
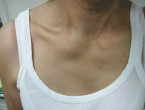


Βότανα-Φυσικές ουσίες
